SUMMARY: Patients undergoing cytotoxic chemotherapy and Hematopoietic Stem Cell Transplantation (HSCT), when neutropenic, are at risk for infection. The risk of infection increases with the depth and duration of neutropenia. The greatest infection risk is among those who experience profound, prolonged neutropenia after chemotherapy. Neutropenia is defined as an absolute neutrophil count of less than 1,000/µL, severe neutropenia as absolute neutrophil count less than 500/µL and profound neutropenia as less than 100/µL. Neutropenia is considered protracted if it lasts for 7 days or more. Fever in neutropenic patients is defined as a single oral temperature of 38.3°C (101°F) or more or a sustained temperature of 38.0°C (100.4°F) or more over a period of 1 hour.
The ASCO in partnership with Infectious Diseases Society of America (IDSA) convened an Expert Panel and updated the 2013 ASCO guideline on antimicrobial prophylaxis for immunosuppressed adult patients undergoing treatment of malignancy. The expert panel conducted a systematic review of relevant studies which included six new or updated meta-analyses and six new primary studies, from May 2011 to November 2016. The guideline recommendations were based on the review of evidence by the Expert Panel.
KEY RECOMMENDATIONS
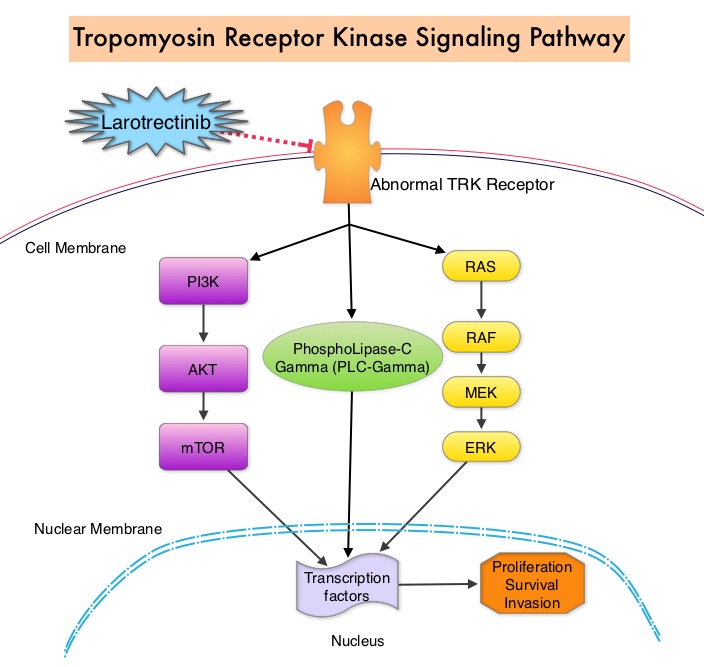
CLINICAL QUESTION 1 – Antibacterial Prophylaxis: Does antibacterial prophylaxis with a Fluoroquinolone, compared with placebo, no intervention, or another class of antibiotic, reduce the incidence of and mortality as a result of febrile episodes in patients with cancer?
Recommendation: Antibiotic prophylaxis with a Fluoroquinolone is recommended for patients who are at high risk for Febrile Neutropenia or profound, protracted neutropenia, such as those patients with Acute Myeloid Leukemia/Myelodysplastic syndromes (AML/MDS) or Hematopoietic Stem Cell Transplantation (HSCT) treated with myeloablative conditioning regimens. Antibiotic prophylaxis is not routinely recommended for patients with solid tumors, for patients who are at low risk of profound, protracted neutropenia and when CSF prophylaxis effectively reduces the severity and duration of neutropenia.
CLINICAL QUESTION 2 – Antifungal Prophylaxis: Does antifungal prophylaxis with an oral triazole or parenteral Echinocandin, compared with no prophylaxis, or another treatment option, reduce the incidence of and mortality, as a result of febrile episodes in patients with cancer?
Recommendation 2.1: Antifungal prophylaxis with an oral triazole or parenteral echinocandin is recommended for patients who are at risk for profound, protracted neutropenia and mucositis, such as most patients with AML/MDS or HSCT. Antifungal prophylaxis is not routinely recommended for patients with solid tumors. Clinicians should be able to differentiate the risks for invasive candidiasis from the risks for invasive mold infection. This is because Fluconazole is active against yeast but not mold whereas Echinocandins and other azole antifungals, such as Posaconozole, Voriconozole, or Isavuconazole are mold-active agents. A mold-active triazole is recommended where the risk of invasive Aspergillosis is more than 6%, such as in patients with AML/MDS during the neutropenic period associated with chemotherapy, in the late stage postallogeneic SCT and/or in the context of GVHD. Antifungal prophylaxis is not routinely recommended for patients who are at low risk of profound, protracted neutropenia and when CSF prophylaxis effectively reduces the severity and duration of neutropenia.
Recommendation 2.2: Prophylaxis is recommended with Trimethoprim-Sulfamethoxazole (TMP-SMX), for patients receiving chemotherapy regimens associated with more than 3.5% risk for pneumonia from Pneumocystis jirovecii (eg, for those on 20 mg or more of prednisone daily for more than 4 weeks). For those hypersensitive to Sulfonamides or unable to tolerate TMP-SMX, alternative options include Dapsone, aerosolized Pentamidine, or Atovaquone.
CLINICAL QUESTION 3 – Antiviral Prophylaxis: Does antiviral prophylaxis reduce the incidence of immunosuppression-related viral infections in patients with cancer compared with no prophylaxis or another treatment option?
Recommendation 3.1: Herpes Simplex Virus-seropositive patients undergoing allogeneic HSCT or leukemia induction therapy should receive prophylaxis with a nucleoside analog such as Acyclovir.
Recommendation 3.2: For patients who are at high risk of Hepatitis B Virus reactivation, treatment with a nucleoside reverse transcription inhibitor (eg, Entecavir or Tenofovir) is recommended.
Recommendation 3.3: Yearly influenza vaccination with inactivated quadrivalent vaccine is recommended for all patients receiving chemotherapy for malignancy and all family and household contacts and health care providers. It is best administered more than 1 week after the last treatment or more than 2 weeks before chemotherapy administration. Individuals older than 65 years should receive the high-dose vaccine.
Recommendation 3.4: The Expert Panel also supports other vaccination recommendations for immunosuppressed adult oncology patients that are contained within the IDSA guideline for vaccination of the immunosuppressed patients.
CLINICAL QUESTION 4 – Do additional precautions, such as hand hygiene, air filtration, or a neutropenic diet, reduce the risk of infection in neutropenic patients with cancer compared with no or other additional precautions?
Recommendation 4.1: All health care workers should comply with hand hygiene and respiratory hygiene/cough etiquette guidelines to reduce the risk for aerosol- and direct or indirect contact-based transmission of pathogenic microorganisms in the health care setting.
Recommendation 4.2: Outpatients with neutropenia from cancer therapy should avoid prolonged contact with environments that have high concentrations of airborne fungal spores (eg, construction and demolition sites, intensive exposure to soil through gardening or digging, or household renovation).
Antimicrobial Prophylaxis for Adult Patients With Cancer-Related Immunosuppression: ASCO and IDSA Clinical Practice Guideline Update. Taplitz RA, Kennedy EB, Bow EJ, et al. J Clin Oncol 2018;36:3043-3054

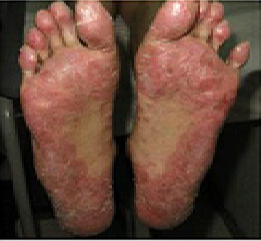
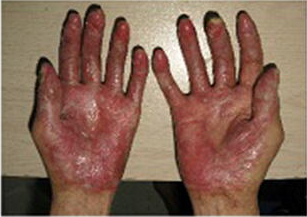 In previously published studies, JAKAFI® when used in patients with refractory GVHD demonstrated an Overall Response Rate of 85% in acute or chronic GVHD, with a 25% Complete Remission rate.
In previously published studies, JAKAFI® when used in patients with refractory GVHD demonstrated an Overall Response Rate of 85% in acute or chronic GVHD, with a 25% Complete Remission rate.