
The FDA on May 27, 2026, approved DECNUPAZ®, a CD123-directed antibody and alkylating agent conjugate, for adults with Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN). DECNUPAZ® is a product of AbbVie, Inc.
Tag: General Medical Oncology & Hematology
ASCO Guideline Update on WBC Growth Factors
SUMMARY: Neutropenia is a clinical challenge and its associated complications, such as febrile neutropenia and infection, are significant toxicities resulting from myelosuppressive chemotherapy. These complications necessitate immediate evaluation and treatment, frequently requiring empirical antibiotics and hospitalization. Hematopoietic Colony-Stimulating Factors (CSFs) are used to decrease the severity and duration of neutropenia and are recommended for patients at high risk of neutropenia due to chemotherapy regimens, age, or comorbidities, thereby reducing the risk of febrile neutropenia. Further, they are also utilized for stem-cell mobilization in transplantation (SCT). ASCO guidelines (updated in 2015) provided evidence-based guidance on the appropriate use of CSFs in adults receiving cancer treatment.
The present ASCO guideline update on use of hematopoietic Colony-Stimulating Factors (CSFs) in cancer patients was based on review of 33 randomized controlled trials and 16 meta-analyses, and systematic reviews, that addressed use of CSFs for the prevention or treatment of neutropenic events, published from September 1, 2014, to August 22, 2025.
Guideline Questions and Recommendations
Clinical Question: What factors should influence the decision to administer primary prophylaxis of febrile neutropenia with a CSF?
Recommendation:
1.1. Patients should be offered primary prophylaxis with a G-CSF when the risk of febrile neutropenia, secondary to a chemotherapy regimen, is equal to or greater than approximately 20%, unless an alternative chemotherapy regimen with comparable efficacy and safety that does not require G-CSF is available.
1.2. Among patients who receive chemotherapy with a lower risk of febrile neutropenia, primary prophylaxis with a G-CSF should be offered if a patient is at high risk of complications from febrile neutropenia based on age, comorbidities, or disease characteristics, and no alternative chemotherapy regimen with comparable efficacy and safety that does not require G-CSF is available.
1.3. If G-CSF is not affordable or available, antibiotic prophylaxis may be offered.
Qualifying statement for Recommendation 1.3: Antibiotic prophylaxis is not a preferred option due to the risk of antimicrobial resistance, disturbance of gut microbiome, Clostridioides difficile infection, and other adverse effects, which may outweigh the benefits of antibiotic use in many cases.
Clinical Question: What factors should influence the decision to administer secondary prophylaxis of febrile neutropenia with a CSF?
Recommendation:
2.1. Secondary prophylaxis with a CSF is recommended for patients who experienced a neutropenic complication from a previous cycle of chemotherapy (for which primary prophylaxis was not received), in which a reduced dose or treatment delay may compromise cure rates or survival outcomes. In many clinical situations, dose reduction or delay may be a reasonable alternative or additional strategy.
Clinical Question: Are there circumstances in which CSFs may be administered for the treatment of febrile neutropenia?
Recommendation:
3.1. A CSF should not be routinely used for patients with neutropenia who are afebrile.
3.2. A CSF should not be routinely used as adjunctive treatment with antibiotic therapy for patients with fever and neutropenia.
3.3. A CSF may be offered in patients with fever and neutropenia who are at high risk for infection-associated complications or who have prognostic factors that are predictive of poor clinical outcomes.
Clinical Question: What is the role of CSFs as adjuncts to progenitor-cell transplantation?
Recommendation:
4.1. A CSF should be used alone, after chemotherapy, or in combination with a CXCR4 inhibitor (Plerixafor or Motixafortide), to mobilize peripheral-blood progenitor cells. Choice of mobilization strategy depends in part on the type of cancer and type of transplantation.
4.2. A CSF should be administered after autologous SCT to reduce the duration of severe neutropenia.
4.3. A CSF may be administered after allogeneic SCT to reduce the duration of severe neutropenia.
Clinical Question: Should CSFs be avoided in patients receiving concomitant chemotherapy and radiation therapy?
Recommendation:
5.1. CSFs are not recommended in patients receiving concomitant chemotherapy and radiation therapy, particularly involving the mediastinum.
Note for Recommendation 5.1: There is little evidence regarding use of CSFs in patients receiving radiation therapy alone.
Clinical Question: Do CSFs differ in efficacy or safety?
Recommendation:
6.1. Filgrastim, Pegfilgrastim, Eflapegrastim, and biosimilars can be used for prophylaxis or treatment of febrile neutropenia. The choice of agent depends on cost, patient convenience, availability, accessibility, health system context, disease subtype, and treatment regimen.
The following factors increase the risk for Febrile Neutropenia in addition to chemotherapy regimen and type of malignancy
- Age 65 years or older or frailty based on geriatric assessment
- Advanced disease
- Previous chemotherapy or radiation therapy
- Preexisting neutropenia or bone marrow involvement with tumor
- Active or recent infection
- Colonization with multidrug-resistant organisms
- Open wounds or recent surgery
- Poor performance status
- Poor nutritional status or low BMI
- Poor renal function
- Liver dysfunction, most notably elevated bilirubin
- Cardiovascular disease
- HIV
- Multiple comorbid conditions
WBC Growth Factors: ASCO Guideline Update. Gyawali B, Bohlke K, Dickter JK, et al. J Clin Oncol. 2026; 44:812-824.
RYBREVANT FASPRO® (Amivantamab and Hyaluronidase-lpuj)
The FDA on December 17, 2025, approved RYBREVANT FASPRO® for subcutaneous injection for adult patients across all indications approved for the intravenous formulation of RYBREVANT®. See the prescribing information for the specific indications. RYBREVANT FASPRO® is a product of Janssen Biotech, Inc.
KOSELUGO® (Selumetinib)
The FDA on November 19, 2025, approved KOSELUGO® for adults with NeuroFibromatosis type 1 (NF1) who have symptomatic, inoperable Plexiform Neurofibromas (PN). FDA previously approved KOSELUGO® capsules and granules for pediatric patients 1 year of age and older for this indication. KOSELUGO® is a product of AstraZeneca Pharmaceuticals LP.
POHERDY® (Pertuzumab-dpzb)
The FDA on November 13, 2025, approved POHERDY® as an interchangeable biosimilar to PERJETA® (Pertuzumab). This is the first approval of a biosimilar for PERJETA®. POHERDY® is a product of Shanghai Henlius Biologics Co. Ltd. and PERJETA® is a product of Genentech Inc.
KEYTRUDA QLEX® (Pembrolizumab and Berahyaluronidase alfa-pmph)
The FDA on September 19, 2025, approved KEYTRUDA QLEX® (Pembrolizumab and Berahyaluronidase alfa-pmph) (Keytruda Qlex, Merck) for subcutaneous injection for adult and pediatric (12 years and older) solid tumor indications approved for the intravenous formulation of pembrolizumab (Keytruda, Merck). See the prescribing information for the specific indications.
KOSELUGO® (Selumetinib)
The FDA on September 10, 2025, approved KOSELUGO® granules and capsules for pediatric patients 1 year of age and older with neurofibromatosis type 1 (NF1) who have symptomatic, inoperable Plexiform Neurofibromas (PN). FDA previously approved KOSELUGO® capsules for pediatric patients 2 years of age and older with NF1 who have symptomatic, inoperable PN. KOSELUGO® is a product of AstraZeneca Pharmaceuticals LP.
Therapeutic Prowess and Potential of Multifunctional Therapeutics: A Review of Bispecific Antibodies
Written by: Jaffer A. Ajani, MD, FASCO
This educational opportunity is sponsored by: Jazz Pharmaceuticals
Concept and Technology
Bispecific antibodies (BsAbs) transcend conventional limitations of therapeutic protein engineering by simultaneously engaging two distinct biological targets. Rooted in molecular cooperation, BsAbs combine two functional antigen-binding fragments (often Fab arms) into a single molecule.1 A considerable novelty over traditional monoclonal antibodies (mAbs), which target a single epitope, BsAbs lead to forced cellular proximity or receptor clustering.1–3 Technological challenges of manufacturing BsAbs for optimal pharmacokinetics (PK), stability, and purity remain. Yet, their dual-targeting allows BsAbs to mediate synergistic effects and intervene in complex, multi-factorial disease pathways—for example, in oncology, where we find multiple redundant receptors, ligands, and evasion mechanisms.1 The following review will review BsAb structures, mechanisms of action, safety profiles, and future directions.
Structural Variants
- Non-IgG-like (Fc-Silent) variants are characterized by a lack of the Fragment crystallizable (Fc) domain, resulting in small molecules that are rapidly cleared by the kidneys, necessitating frequent dosing. Their advantage is high potency and efficient tissue penetration.4 These include:
- Bispecific T-Cell Engagers (BiTEs): Typically constructed as tandem single-chain variable fragments (scFv) that link a tumor-associated antigen (TAA) binder and a CD3 binder via a peptide linker.4 Blinatumomab is a well-known example that achieves potent cellular redirection.
- Dual Affinity Re-targeting Molecules (DARTs): Similar to BiTEs, DARTs incorporate an additional disulfide bridge to improve structural stability.
- Killer Cell Engagers (BiKEs/TriKEs): These target the innate immune system by engaging CD16 on NK cells. Trispecific Killer Engagers (TriKEs) feature a third component, such as an IL-15 crosslinker, to sustain NK cell proliferation and cytotoxicity.4
- IgG-like (Fc-Containing) formats retain the Y-shaped IgG structure, including the Fc domain, conferring prolonged serum half-life via FcRn recycling.4 However, assembling two different heavy chains and two different light chains into a functional heterodimer without forming undesirable mispaired byproducts demands intensive engineering—e.g., CrossMab and/or Knobs-into-Holes (KiH) technologies.4–6
Mechanisms of Action (MOA)
The therapeutic power of BsAbs lies in their ability to execute mechanisms categorized as acting in-trans or in-cis, based on their molecular or cellular target configuration.
- In-Trans Mechanisms: The core in-trans function is creating a physical linkage between two distinct molecular or cellular entities. These include:
- Cellular Bridging (T-Cell Engagers; TCEs): This is the hallmark of oncology BsAbs. By simultaneously binding a TAA and CD3 on T cells, the BsAb forces a physical link, forming a cytolytic synapse.6 This mechanism bypasses the need for natural T-cell receptor (TCR) clustering and Major Histocompatibility Complex (MHC) presentation, allowing the T cell to attack regardless of the tumor’s MHC status.4,6
- Co-factor Mimicry: Outside of cytotoxicity, BsAbs can direct components to form a functional complex. Emicizumab, approved for Hemophilia A, is an example.2–6
- In-Cis Mechanisms: Involve targeting components that reside on the same cell or act within the same signaling pathway. These include:
- Dual Signaling Inhibition (Dual Blockade): Simultaneously blocks two different receptors or ligands to suppress synergistic pathways crucial for disease progression.
- Examples: Targeting HER2/HER3 (Zenocutuzumab) or EGFR/MET (Amivantamab) to halt parallel proliferation cascades in cancer.1–6
- Biparatopic Engagement: By binding two distinct, non-overlapping epitopes on the same antigen4,5, biparatopics intensify control over one oncogenic “addiction” pathway via geometry-driven clustering, internalization, and boosted Fc effector functions. Biparatopics enhance binding avidity and promote superior functional modulation of the target, such as forced receptor clustering and internalization, the latter being highly beneficial for Antibody-Drug Conjugates (ADCs). Biparatopic binding drives dense clustering of the same receptor, leading to “caps” on the cell surface, resulting in potent receptor internalization and degradation. This yields deeper and more durable signal blockade than a single monoclonal antibody or cocktail.7 The high local receptor and antibody density also enables multimodal effector functions, and helps overcome resistance within a single pathway by engaging distinct functional domains to block both ligand-dependent and ligand-independent signaling and interfere with heterodimerization (e.g., HER2/HER3). They also retain efficacy when tumors escape mono-epitope antibodies through epitope masking or mutation.
- Example: Zanidatamab, which targets two distinct HER2 epitopes, and is unique in its ability to induce receptor clustering and “capping.”8-9
- Dual Signaling Inhibition (Dual Blockade): Simultaneously blocks two different receptors or ligands to suppress synergistic pathways crucial for disease progression.
Clinical Landscape
The BsAb landscape has rapidly expanded since the first approval of Blinatumomab in 2014, reflecting a growing therapeutic impact across multiple disease areas. As of late 2025, fifteen bispecific molecules have secured FDA approval, spanning both oncology and non-oncology indications. This surge underscores the versatility of BsAbs and their ability to address complex biological pathways through innovative mechanisms of action.
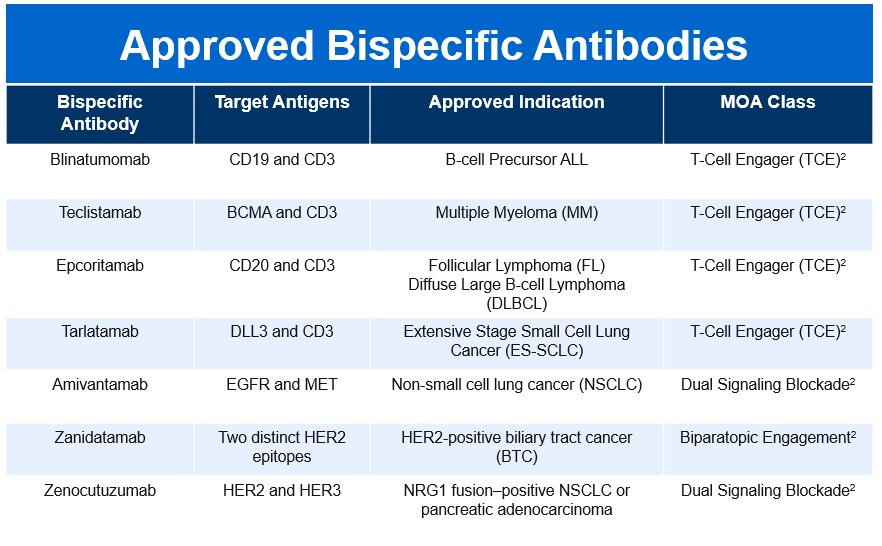
Limitations, Safety, and Risk Mitigation
- Manufacturing Challenges: The inherent complexity of BsAbs introduces challenges related to stability, manufacturability, and impurity control.1 The fusion of exogenous antigen-binding domains can decrease biophysical stability, and the complex assembly process frequently results in the formation of product-related impurities and mispaired species, which are difficult to remove during purification. These factors are not merely manufacturing hurdles; they directly influence the biological activity and, critically, the immunogenic potential of the final drug product.
- MOA-Specific Toxicity: The safety profile of BsAbs is highly dependent on their MOA
- T-Cell Engager Toxicity: The highly potent, acute T-cell activation triggered by TCEs results in two major, distinct safety concerns. The first is Cytokine Release Syndrome (CRS), a serious acute toxicity caused by the mass release of systemic cytokines. CRS has been reported in up to 70% of patients receiving BsAbs, often necessitating hospitalization and precise management protocols. Severe cases (Grade ≥ 3) occur in 5–10% of patients.6 The second concern is Neurotoxicity (ICANS), which, while less frequent than CRS, affects 10–15% of patients and can range from mild confusion to cerebral edema.6 In addition, there is an Immune Regulation Paradox. Paradoxically, T-BsAb therapy can trigger the expansion and activation of inhibitory Regulatory T (Treg) cells in the tumor microenvironment, leading to the production of anti-inflammatory cytokines like IL-10. This critically inhibits the desired effector T-cell response, suggesting that combination strategies—such as transient Treg ablation—may be necessary to maximize efficacy.6
- Pathway Blocker and Biparatopic Toxicity: These agents generally do not induce acute, systemic cytokine surges. Instead, their adverse event profiles reflect the targeted receptors. For example, the dual signaling blocker Amivantamab (targets EGFR/MET) exhibits EGFR-inhibition-related dermatologic toxicities, like paronychia, skin fissures, and pruritus, as well as infusion reactions. Dual checkpoint inhibitors can have classic IO toxicities. Biparatopic antibodies, like Zanidatamab, demonstrate a manageable profile but frequently cause gastrointestinal toxicities (such as diarrhea and nausea/vomiting) and infusion-related reactions (IRRs).9 Importantly, clinical data for Zanidatamab confirmed no reports of CRS.9
- Mitigating Immunogenicity Risk: The complex structures, engineered sequences, and immunostimulatory MOAs of oncology BsAbs contribute to an increased risk of immunogenicity compared to mAbs. Mitigation must begin at the engineering stage, utilizing in silico prediction and in vitro assays to guide the selection of low-risk antibody constructs through deimmunization and tolerization methods.
Future Directions
The BsAb pipeline remains robust, reflecting a continuous drive toward addressing current clinical limitations and expanding into novel biological territories. The future of BsAbs is characterized by a strategic shift toward overcoming the immunosuppressive tumor microenvironment (TME). Emerging candidates are now focused on targets that modulate the innate immune system and TME suppression, such as LILRB1/2 bispecific IgG1 antibodies for advanced solid tumors.10-11 Furthermore, BsAbs are expanding beyond simple blockade, with molecules like SAR446422 (CD28xOX40 bispecific) in trial for inflammatory indications, demonstrating the potential for BsAbs to achieve synergistic co-stimulatory agonism.10-11 The continuous innovation in structural design, focused now on minimizing impurity-driven immunogenicity and maximizing the therapeutic window, ensures that BsAbs are poised to become the standard for highly tailored, multifunctional therapeutic intervention across diverse and complex diseases. The future of BsAbs is very promising.10-11
References:
- Shan KS, Musleh Ud Din S, Dalal S, Gonzalez T, Dalal M, Ferraro P, Hussein A, Vulfovich M. Bispecific Antibodies in Solid Tumors: Advances and Challenges. International Journal of Molecular Sciences. 2025; 26(12):5838. https://doi.org/10.3390/ijms26125838.
- The Bispecific 2024 Landscape Review. Beacon Intelligence. 2024. https://beacon-intelligence.com/landscape-reviews/bispecific/. Accessed November 23, 2025.
- Ai Z, Wang B, Song Y, Cheng P, Liu X, Sun P. Prodrug-based bispecific antibodies for cancer therapy: advances and future directions. Front Immunol. 2025;16:1523693. Published 2025 Jan 22. doi:10.3389/fimmu.2025.1523693.
- Amash A, Volkers G, Farber P, et al. Developability considerations for bispecific and multispecific antibodies. MAbs. 2024;16(1):2394229. doi:10.1080/19420862.2024.2394229.
- Shui L, Wu D, Yang K, Sun C, Li Q, Yin R. Bispecific antibodies: unleashing a new era in oncology treatment. Mol Cancer. 2025;24(1):212. Published 2025 Aug 4. doi:10.1186/s12943-025-02390-y.
- Dewaele L, Fernandes RA. Bispecific T-cell engagers for the recruitment of T cells in solid tumors: a literature review. Immunother Adv. 2025;5(1):ltae005. Published 2025 Jan 27. doi:10.1093/immadv/ltae005.
- Kast F, Schwill M, Stüber JC, et al. Engineering an anti-HER2 biparatopic antibody with a multimodal mechanism of action. Nat Commun. 2021;12(1):3790. Published 2021 Jun 18. doi:10.1038/s41467-021-23948-6.
- Elimova E, Ajani J, Burris H, et al. Zanidatamab plus chemotherapy as first-line treatment for patients with HER2-positive advanced gastro-oesophageal adenocarcinoma: primary results of a multicentre, single-arm, phase 2 study. Lancet Oncol. 2025;26(7):847-859. doi:10.1016/S1470-2045(25)00287-6.
- Ziihera Safety Information. Ziihera HCP (Jazz Pharmaceuticals). https://www.ziiherahcp.com/safety. Accessed November 23, 2025.
- Wen J, Cui W, Yin X, et al. Application and future prospects of bispecific antibodies in the treatment of non-small cell lung cancer. Cancer Biol Med. 2025;22(4):348-375. doi:10.20892/j.issn.2095-3941.2024.0470.
- Engineering the Next Generation of Bispecific Antibodies. PEGS Europe 2024 Archive. https://www.pegsummiteurope.com/24/engineering-bispecifics. Accessed November 23, 2025
Precision Medicine in Practice: Timely Use of Tumor NGS Remains Suboptimal in Common Cancers
SUMMARY: Next-generation sequencing (NGS) has revolutionized the management of advanced cancers by enabling identification of tumor-specific genomic alterations for which targeted therapies are now available. National guidelines recommend early and routine NGS testing for patients with advanced or metastatic solid tumors to inform treatment decisions. In the United States, the five most prevalent advanced or metastatic solid tumors include advanced Non-Small Cell Lung Cancer (aNSCLC), metastatic Breast Cancer (mBC), metastatic Prostate Cancer (mPC), advanced Colorectal Cancer (aCRC), and metastatic Pancreatic Cancer (mPanC). For these malignancies, the integration of NGS has become increasingly critical in guiding targeted therapy selection and improving survival outcomes. Despite the approval of multiple targeted therapies for these malignancies, real-world utilization of NGS remains inconsistent.
In this study presented at the 2025 ASCO Annual Meeting, Chehade and colleagues, evaluated patterns in NGS testing and its timing, relative to patient mortality.
Study Overview: This retrospective analysis leveraged the Flatiron Health EHR-derived de-identified database across 280 cancer clinics, spanning data from 2011 onward. The study included patients with a diagnosis of aNSCLC, mBC, mPC, aCRC, or mPanC, all of whom had records of NGS testing and a documented date of death. The researchers identified 86,536 patients with advanced non-small cell lung cancer, 36,000 with metastatic breast cancer, 35,702 with advanced colorectal cancer, 24,105 with metastatic prostate cancer and 14,964 with metastatic pancreatic cancer. About a third of patients from each cancer group received NGS testing (NSCLC, 36.3%; breast cancer, 32.1%; colorectal cancer, 41%; prostate cancer, 30.9%; and pancreatic cancer, 35.4%).
Patients were categorized based on the interval between receipt of NGS results and death:
- More than 3 months before death
- Within 3 months of death
- After death
Key Findings Across cancer types, only 30% to 40% of patients received NGS testing. Among those who were tested and had a recorded date of death, the timing of NGS was as follows:
| Timing of First NGS | aNSCLC (N=19,958) | mBC (N=5,689) | mPC (N=3,397) | aCRC (N=8,553) | mPanC (N=3,957) |
| >3 mo before death | 72.3% | 81.6% | 85.4% | 85.0% | 71.1% |
| Within 3 mo of death | 25.6% | 16.9% | 13.5% | 13.7% | 26.5% |
| After death | 2.1% | 1.5% | 1.1% | 1.3% | 2.4% |
Notably, up to one in four patients with NSCLC or pancreatic cancer received their first NGS results within 3 months of death, a timeframe often too late for actionable therapeutic intervention.
Interpretation and Implications Despite advances in molecularly targeted therapies and growing guideline support for comprehensive genomic profiling, real-world testing patterns remain suboptimal:
- Low uptake: Only about a third of eligible patients undergo NGS testing.
- Late testing: A substantial proportion of tested patients receive results within 3 months of death.
- Missed opportunities: Many patients are never tested—or are tested too late to benefit from life-extending therapies.
These findings highlight ongoing gaps in precision oncology implementation, especially in community-based settings.
Next Steps & Recommendations To improve the utility of NGS in oncology, efforts should focus on:
- Earlier testing: At diagnosis or at first progression of advanced disease.
- Workflow integration: Embedding NGS into routine clinical pathways.
- Education: Raising awareness among clinicians and patients about the benefits of timely testing.
- Health system support: Addressing barriers such as reimbursement, turnaround times, and tissue availability.
Conclusion: Real-World Data from this large retrospective analysis reveal late-stage testing and underutilization of life-prolonging genomic profiling. This study underscores an urgent need to optimize the timing and uptake of NGS testing in patients with advanced solid tumors. Earlier and broader testing is essential to ensure patients have access to the most effective, personalized treatment strategies, and to avoid the missed potential of life-extending therapies.
Utilization and timing of first tumor next-generation sequencing testing (NGS) in patients (pts) with five most common cancers in the USA. Chehade CH, Jo Y, Ozay ZI, et al. Doi: 10.1200/JCO.2025.43.16_suppl.11014. Abstract # 11014. Presented at: ASCO Annual Meeting; May 30-June 3, 2025; Chicago.
Precision Medicine in Practice: Timely Use of Tumor NGS Remains Suboptimal in Common Cancers
SUMMARY: Next-generation sequencing (NGS) has revolutionized the management of advanced cancers by enabling identification of tumor-specific genomic alterations for which targeted therapies are now available. National guidelines recommend early and routine NGS testing for patients with advanced or metastatic solid tumors to inform treatment decisions. In the United States, the five most prevalent advanced or metastatic solid tumors include advanced Non-Small Cell Lung Cancer (aNSCLC), metastatic Breast Cancer (mBC), metastatic Prostate Cancer (mPC), advanced Colorectal Cancer (aCRC), and metastatic Pancreatic Cancer (mPanC). For these malignancies, the integration of NGS has become increasingly critical in guiding targeted therapy selection and improving survival outcomes. Despite the approval of multiple targeted therapies for these malignancies, real-world utilization of NGS remains inconsistent.
In this study presented at the 2025 ASCO Annual Meeting, Chehade and colleagues, evaluated patterns in NGS testing and its timing, relative to patient mortality.
Study Overview: This retrospective analysis leveraged the Flatiron Health EHR-derived de-identified database across 280 cancer clinics, spanning data from 2011 onward. The study included patients with a diagnosis of aNSCLC, mBC, mPC, aCRC, or mPanC, all of whom had records of NGS testing and a documented date of death. The researchers identified 86,536 patients with advanced non-small cell lung cancer, 36,000 with metastatic breast cancer, 35,702 with advanced colorectal cancer, 24,105 with metastatic prostate cancer and 14,964 with metastatic pancreatic cancer. About a third of patients from each cancer group received NGS testing (NSCLC, 36.3%; breast cancer, 32.1%; colorectal cancer, 41%; prostate cancer, 30.9%; and pancreatic cancer, 35.4%).
Patients were categorized based on the interval between receipt of NGS results and death:
- More than 3 months before death
- Within 3 months of death
- After death
Key Findings Across cancer types, only 30% to 40% of patients received NGS testing. Among those who were tested and had a recorded date of death, the timing of NGS was as follows:
| Timing of First NGS | aNSCLC (N=19,958) | mBC (N=5,689) | mPC (N=3,397) | aCRC (N=8,553) | mPanC (N=3,957) |
| >3 mo before death | 72.3% | 81.6% | 85.4% | 85.0% | 71.1% |
| Within 3 mo of death | 25.6% | 16.9% | 13.5% | 13.7% | 26.5% |
| After death | 2.1% | 1.5% | 1.1% | 1.3% | 2.4% |
Notably, up to one in four patients with NSCLC or pancreatic cancer received their first NGS results within 3 months of death, a timeframe often too late for actionable therapeutic intervention.
Interpretation and Implications Despite advances in molecularly targeted therapies and growing guideline support for comprehensive genomic profiling, real-world testing patterns remain suboptimal:
- Low uptake: Only about a third of eligible patients undergo NGS testing.
- Late testing: A substantial proportion of tested patients receive results within 3 months of death.
- Missed opportunities: Many patients are never tested—or are tested too late to benefit from life-extending therapies.
These findings highlight ongoing gaps in precision oncology implementation, especially in community-based settings.
Next Steps & Recommendations To improve the utility of NGS in oncology, efforts should focus on:
- Earlier testing: At diagnosis or at first progression of advanced disease.
- Workflow integration: Embedding NGS into routine clinical pathways.
- Education: Raising awareness among clinicians and patients about the benefits of timely testing.
- Health system support: Addressing barriers such as reimbursement, turnaround times, and tissue availability.
Conclusion: Real-World Data from this large retrospective analysis reveal late-stage testing and underutilization of life-prolonging genomic profiling. This study underscores an urgent need to optimize the timing and uptake of NGS testing in patients with advanced solid tumors. Earlier and broader testing is essential to ensure patients have access to the most effective, personalized treatment strategies, and to avoid the missed potential of life-extending therapies.
Utilization and timing of first tumor next-generation sequencing testing (NGS) in patients (pts) with five most common cancers in the USA. Chehade CH, Jo Y, Ozay ZI, et al. Doi: 10.1200/JCO.2025.43.16_suppl.11014. Abstract # 11014. Presented at: ASCO Annual Meeting; May 30-June 3, 2025; Chicago.