*3-5–minute vs 30-minute infusion of IV nivolumab. This does not account for all aspects of treatment. Actual clinic time may vary. 1,2
Expert opinion: Saby George, MD, FACP†
†Dr Saby George, MD, is a paid consultant of Bristol Myers Squibb (BMS) who was compensated by BMS for his contributions to this article.
Content sponsored by Bristol Myers Squibb
Subcutaneous administration overview
While immune checkpoint inhibitors have emerged as key treatment options for certain types of cancer, they are primarily delivered through intravenous (IV) administration,3 creating a need for alternative routes of administration.4-5 A subcutaneous (SC) injection may reduce the time preparing and administering treatment compared to IV delivery, offer practice flexibility that may free up infusion chairs, and deliver treatment faster.3,5 “Infusion centers are overwhelmed. Infusion chairs may open up if we transition to approved SC options,” remarked Dr George.
Evaluation of comparable PK, efficacy, and safety of SC OPDIVO Qvantig with IV nivolumab
OPDIVO Qvantig is formulated with hyaluronidase to increase the dispersion and absorption of SC nivolumab.1 CheckMate 67T, a randomized, open-label, phase 3 noninferiority trial, was designed to compare the PK, efficacy, and safety of OPDIVO Qvantig (delivered as a SC injection) with IV nivolumab.1,4
OPDIVO QVANTIG, as monotherapy, is indicated for the first-line treatment of adult patients with intermediate- or poor-risk advanced renal cell carcinoma (RCC), following treatment with intravenous nivolumab and ipilimumab combination therapy. OPDIVO QVANTIG is not indicated in combination with ipilimumab for the treatment of renal cell carcinoma. Please see additional 16 indications below.
OPDIVO QVANTIG is associated with the following Warnings and Precautions: severe and fatal immune-mediated adverse reactions including pneumonitis, colitis, hepatitis and hepatotoxicity, endocrinopathies, nephritis with renal dysfunction, dermatologic adverse reactions, other immune-mediated adverse reactions; complications of allogeneic hematopoietic stem cell transplantation (HSCT); embryo-fetal toxicity; and increased mortality in patients with multiple myeloma when OPDIVO QVANTIG is added to a thalidomide analogue and dexamethasone, which is not recommended outside of controlled clinical trials.
Please see Important Safety Information for OPDIVO QVANTIG below and US Full Prescribing Information for OPDIVO QVANTIG.
CheckMate 67T was a phase 3, randomized (1:1), open-label, noninferiority trial evaluating OPDIVO Qvantig (1,200 mg of nivolumab and 20,000 units of hyaluronidase) compared to intravenous nivolumab, in adult patients with advanced or metastatic clear-cell renal cell carcinoma (ccRCC) who received prior systemic therapy.1,4 Patients were stratified by weight (<80 kg versus ≥80 kg) and International Metastatic RCC Database Consortium (IMDC) risk score (favorable vs intermediate vs poor risk).1 A total of 495 patients were randomized to receive either OPDIVO Qvantig every 4 weeks subcutaneously (n=248) or nivolumab 3 mg/kg every 2 weeks intravenously (n=247).1,4 The co-primary endpoints were time-averaged serum concentration over 28 days (Cavgd28) and minimum serum concentration at steady state (Cminss).4 The key powered secondary endpoint was overall response rate, as assessed by blinded independent central review. The minimum follow-up time was 8 months.4
Pharmacokinetic, efficacy, and safety results
CheckMate 67T demonstrated that the PK of OPDIVO Qvantig was noninferior to that of intravenously administered nivolumab.1,4*

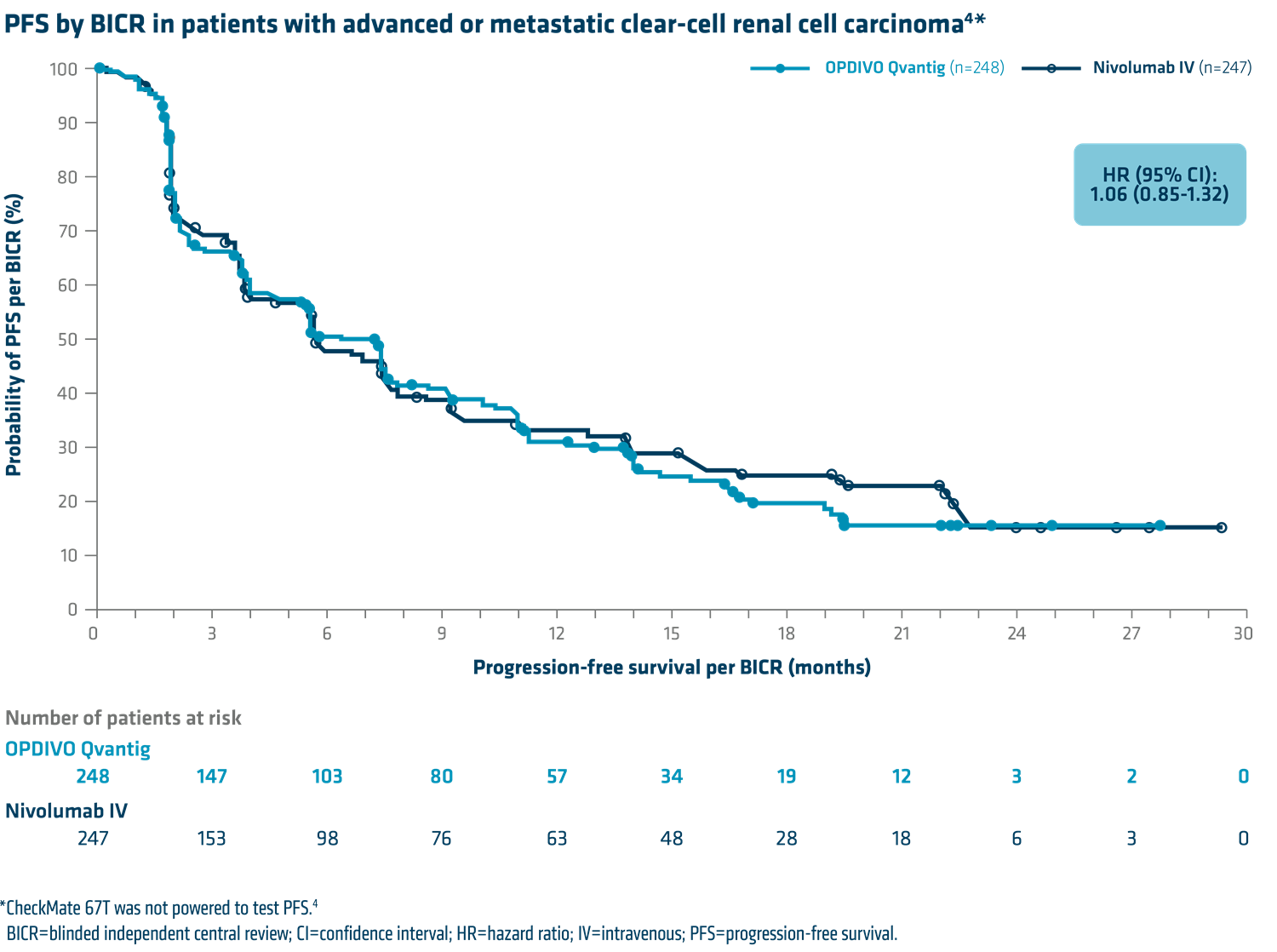
OPDIVO Qvantig resulted in a safety profile comparable with IV nivolumab.1 Dr George noted, “Safety was similar between administration methods. Rates of adverse reactions were similar for IV and SC nivolumab administration.”6 Please see safety table below for more information.

Summary and conclusions
OPDIVO Qvantig resulted in comparable PK, efficacy, and safety to IV nivolumab and may be the right option for your eligible patients.1 This 3-5 minute SC injection option may reduce the steps required for preparation and time needed for administration compared to IV nivolumab.1,2* There is no need for IV preparation, dilution, weight-based dose calculations, or port access with OPDIVO Qvantig.1 According to Dr George, “For my appropriate patients, it gives me flexibility. It may save administration time.* For eligible patients, it’s great to have this subcutaneous treatment option.”
*3-5–minute vs 30-minute infusion of IV nivolumab. This does not account for all aspects of treatment. Actual clinic time may vary.1,2
INDICATIONS
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the first-line treatment of adult patients with intermediate- or poor-risk advanced renal cell carcinoma (RCC), following treatment with intravenous nivolumab and ipilimumab combination therapy.
Limitations of Use: OPDIVO QVANTIG is not indicated in combination with ipilimumab for the treatment of renal cell carcinoma.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), in combination with cabozantinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the treatment of adult patients with unresectable or metastatic melanoma.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the treatment of adult patients with unresectable or metastatic melanoma following treatment with intravenous nivolumab and ipilimumab combination therapy.
Limitations of Use: OPDIVO QVANTIG is not indicated in combination with ipilimumab for treatment of unresectable or metastatic melanoma.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the adjuvant treatment of adult patients with completely resected Stage IIB, Stage IIC, Stage III, or Stage IV melanoma.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), in combination with platinum-doublet chemotherapy, is indicated as neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC).
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), in combination with platinum-doublet chemotherapy, is indicated for the neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC) and no known epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) rearrangements, followed by OPDIVO QVANTIG as monotherapy in the adjuvant setting after surgical resection.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO QVANTIG.
Limitations of Use: OPDIVO QVANTIG is not indicated in combination with ipilimumab for the treatment of metastatic NSCLC.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the treatment of adult patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the adjuvant treatment of adult patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), in combination with cisplatin and gemcitabine, is indicated for the first-line treatment of adult patients with unresectable or metastatic urothelial carcinoma (UC).
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (UC) who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in adult patients who have received neoadjuvant
chemoradiotherapy (CRT).
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC) whose tumors express PD-L1 (≥1%).
Limitations of Use: OPDIVO QVANTIG is not indicated in combination with ipilimumab for the treatment of patients with unresectable advanced or metastatic ESCC.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), as monotherapy, is indicated for the treatment of adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine-and platinum-based chemotherapy.
OPDIVO QVANTIG™ (nivolumab and hyaluronidase-nvhy), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the treatment of adult patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma whose tumors express PD-L1 (≥1%).
IMPORTANT SAFETY INFORMATION
Severe and Fatal Immune-Mediated Adverse Reactions
- Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue. While immune-mediated adverse reactions usually manifest during treatment, they can also occur after discontinuation of OPDIVO QVANTIG. Early identification and management are essential to ensure safe use of OPDIVO QVANTIG. Monitor for signs and symptoms that may be clinical manifestations of underlying immune-mediated adverse reactions. Evaluate clinical chemistries including liver enzymes, creatinine, and thyroid function at baseline and periodically during treatment. In cases of suspected immune-mediated adverse reactions, initiate appropriate workup to exclude alternative etiologies, including infection. Institute medical management promptly, including specialty consultation as appropriate.
- Withhold or permanently discontinue OPDIVO QVANTIG depending on severity (please see Section 2 Dosage and Administration in the accompanying Full Prescribing Information). In general, if OPDIVO QVANTIG interruption or discontinuation is required, administer systemic corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent) until improvement to Grade 1 or less. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over for at least 1 month. Consider administration of other systemic immunosuppressants in patients whose immune-mediated adverse reactions are not controlled with corticosteroid therapy.
- Toxicity management guidelines for adverse reactions that do not necessarily require systemic steroids (e.g., endocrinopathies and dermatologic reactions) are discussed below.
Immune-Mediated Pneumonitis
- OPDIVO QVANTIG can cause immune-mediated pneumonitis. The incidence of pneumonitis is higher in patients who have received prior thoracic radiation.
- Immune-mediated pneumonitis occurred in 2.8% (7/247) of patients receiving OPDIVO QVANTIG, including Grade 3 (0.8%) and Grade 2 (2.0%) adverse reactions.
Immune-Mediated Colitis
- OPDIVO QVANTIG can cause immune-mediated colitis. A common symptom included in the definition of colitis was diarrhea. Cytomegalovirus (CMV) infection/reactivation has been reported in patients with corticosteroid-refractory immune-mediated colitis. In cases of corticosteroid-refractory colitis, consider repeating infectious workup to exclude alternative etiologies.
- Immune-mediated colitis occurred in 2.8% (7/247) of patients receiving OPDIVO QVANTIG, including Grade 3 (0.4%) and Grade 2 (2.4%) adverse reactions.
Immune-Mediated Hepatitis and Hepatotoxicity
- OPDIVO QVANTIG can cause immune-mediated
- Immune-mediated hepatitis occurred in 2.4% (6/247) of patients receiving OPDIVO QVANTIG, including Grade 3 (1.6%), and Grade 2 (0.8%) adverse reactions. Intravenous nivolumab in combination with cabozantinib can cause hepatic toxicity with higher frequencies of Grade 3 and 4 ALT and AST elevations compared to intravenous nivolumab alone. Consider more frequent monitoring of liver enzymes as compared to when the drugs are administered as single agents. With the combination of intravenous nivolumab and cabozantinib, Grades 3 and 4 increased ALT or AST were seen in 11% (35/320) of patients.
Immune-Mediated Endocrinopathies
- OPDIVO QVANTIG can cause primary or secondary adrenal insufficiency, immune-mediated hypophysitis, immune-mediated thyroid disorders, and Type 1 diabetes mellitus, which can present with diabetic ketoacidosis. Withhold OPDIVO QVANTIG depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information). For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically Hypophysitis can present with acute symptoms associated with mass effect such as headache, photophobia, or visual field defects. Hypophysitis can cause hypopituitarism; initiate hormone replacement as clinically indicated. Thyroiditis can present with or without endocrinopathy. Hypothyroidism can follow hyperthyroidism; initiate hormone replacement or medical management as clinically indicated. Monitor patients for hyperglycemia or other signs and symptoms of diabetes; initiate treatment with insulin as clinically indicated.
- Adrenal insufficiency occurred in 2% (5/247) of patients receiving OPDIVO QVANTIG, including Grade 3 (0.8%) and Grade 2 (1.2%) adverse Adrenal insufficiency occurred in 4.7% (15/320) of patients with RCC who received intravenous nivolumab with cabozantinib, including Grade 3 (2.2%) and Grade 2 (1.9%) adverse reactions. Hypophysitis occurred in 0.6% (12/1994) of patients treated with single agent intravenous nivolumab, including Grade 3 (0.2%) and Grade 2 (0.3%). Thyroiditis occurred in 0.4% (1/247) of patients receiving OPDIVO QVANTIG, including a Grade 1 (0.4%) adverse reaction.
- Hyperthyroidism occurred in 0.8% (2/247) of patients receiving OPDIVO QVANTIG, including Grade 2 (0.4%) adverse reactions. Hypothyroidism occurred in 9% (23/247) of patients receiving OPDIVO QVANTIG, including Grade 2 (5.7%) adverse reactions.
- Grade 3 diabetes occurred in 4% (1/247) of patients receiving OPDIVO QVANTIG.
Immune-Mediated Nephritis with Renal Dysfunction
- OPDIVO QVANTIG can cause immune-mediated
- Grade 2 immune-mediated nephritis and renal dysfunction occurred in 1.2% (3/247) of patients receiving OPDIVO QVANTIG.
Immune-Mediated Dermatologic Adverse Reactions
- OPDIVO QVANTIG can cause immune-mediated rash or dermatitis. Exfoliative dermatitis, including Stevens-Johnson Syndrome, toxic epidermal necrolysis (TEN), and DRESS (drug rash with eosinophilia and systemic symptoms), has occurred with PD-1/PD-L1 blocking antibodies. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate non-exfoliative Withhold or permanently discontinue OPDIVO QVANTIG depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information).
- Immune-mediated rash occurred in 7% (17/247) of patients, including Grade 3 (0.8%) and Grade 2 (2.8%) adverse reactions.
Other Immune-Mediated Adverse Reactions
- The following clinically significant immune-mediated adverse reactions occurred at an incidence of <1% (unless otherwise noted) in patients who received OPDIVO QVANTIG or intravenous nivolumab as single agent or in combination with chemotherapy or immunotherapy, or were reported with the use of other PD-1/PD-L1 blocking antibodies. Severe or fatal cases have been reported for some of these adverse reactions: cardiac/vascular: myocarditis, pericarditis, vasculitis; nervous system: meningitis, encephalitis, myelitis and demyelination, myasthenic syndrome/myasthenia gravis (including exacerbation), Guillain-Barré syndrome, nerve paresis, autoimmune neuropathy; ocular: uveitis, iritis, and other ocular inflammatory toxicities can occur; gastrointestinal: pancreatitis to include increases in serum amylase and lipase levels, gastritis, duodenitis; musculoskeletal and connective tissue: myositis/polymyositis, rhabdomyolysis, and associated sequelae including renal failure, arthritis, polymyalgia rheumatica; endocrine: hypoparathyroidism; other (hematologic/immune): hemolytic anemia, aplastic anemia, hemophagocytic lymphohistiocytosis (HLH), systemic inflammatory response syndrome, histiocytic necrotizing lymphadenitis (Kikuchi lymphadenitis), sarcoidosis, immune thrombocytopenic purpura, solid organ transplant rejection, other transplant (including corneal graft) rejection.
- Some ocular IMAR cases can be associated with retinal detachment. Various grades of visual impairment, including blindness, can occur. If uveitis occurs in combination with other immune-mediated adverse reactions, consider a Vogt-Koyanagi-Harada–like syndrome, as this may require treatment with systemic corticosteroids to reduce the risk of permanent vision loss.
Complications of Allogeneic Hematopoietic Stem Cell Transplantation
- Fatal and other serious complications can occur in patients who receive allogeneic hematopoietic stem cell transplantation (HSCT) before or after being treated with OPDIVO QVANTIG. Transplant-related complications include hyperacute graft-versus-host disease (GVHD), acute GVHD, chronic GVHD, hepatic veno-occlusive disease (VOD) after reduced intensity conditioning, and steroid-requiring febrile syndrome (without an identified infectious cause). These complications may occur despite intervening therapy between OPDIVO QVANTIG and allogeneic HSCT.
- Follow patients closely for evidence of transplant-related complications and intervene promptly. Consider the benefit versus risks of treatment with OPDIVO QVANTIG prior to or after an allogeneic HSCT.
Embryo-Fetal Toxicity
- Based on its mechanism of action and data from animal studies, OPDIVO QVANTIG can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of nivolumab to cynomolgus monkeys from the onset of organogenesis through delivery resulted in increased abortion and premature infant Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with OPDIVO QVANTIG and for 5 months after the last dose.
Increased Mortality in Patients with Multiple Myeloma when Nivolumab Is Added to a Thalidomide Analogue and Dexamethasone
- In randomized clinical trials in patients with multiple myeloma, the addition of a PD-1 blocking antibody, including intravenous nivolumab, to a thalidomide analogue plus dexamethasone, a use for which no PD-1 or PD-L1 blocking antibody is indicated, resulted in increased Treatment of patients with multiple myeloma with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials.
Lactation
- There are no data on the presence of nivolumab or hyaluronidase in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment and for 5 months after the last dose of OPDIVO Qvantig.
Serious Adverse Reactions
- In Checkmate 67T, serious adverse reactions occurred in 28% of patients who received OPDIVO QVANTIG (n=247). Serious adverse reactions in >1% of patients included pleural effusion (1.6%), pneumonitis (1.6%), hyperglycemia (1.2%), hyperkalemia (1.2%), hemorrhage (1.2%) and diarrhea (1.2%). Fatal adverse reactions occurred in 3 patients (1.2%) who received OPDIVO QVANTIG and included myocarditis, myositis, and colitis complications. In Checkmate 037, serious adverse reactions occurred in 41% of patients receiving intravenous nivolumab (n=268). Grade 3 and 4 adverse reactions occurred in 42% of patients receiving intravenous nivolumab. The most frequent Grade 3 and 4 adverse drug reactions reported in 2% to <5% of patients receiving intravenous nivolumab were abdominal pain, hyponatremia, increased aspartate aminotransferase, and increased lipase. In Checkmate 066, serious adverse reactions occurred in 36% of patients receiving intravenous nivolumab (n=206). Grade 3 and 4 adverse reactions occurred in 41% of patients receiving intravenous The most frequent Grade 3 and 4 adverse reactions reported in ≥2% of patients receiving intravenous nivolumab were gamma-glutamyltransferase increase (3.9%) and diarrhea (3.4%). In Checkmate 067, the most frequent (≥10%) serious adverse reactions in the intravenous nivolumab arm (n=313) were diarrhea (2.2%), colitis (1.9%), and pyrexia (1.0%). In Checkmate 067, serious adverse reactions (74% and 44%), adverse reactions leading to permanent discontinuation (47% and 18%) or to dosing delays (58% and 36%), and Grade 3 or 4 adverse reactions (72% and 51%) all occurred more frequently in the intravenous nivolumab plus intravenous ipilimumab arm (n=313) relative to the intravenous nivolumab arm (n=313). The most frequent (≥10%) serious adverse reactions in the intravenous nivolumab plus intravenous ipilimumab arm and the intravenous nivolumab arm, respectively, were diarrhea (13% and 2.2%), colitis (10% and 1.9%), and pyrexia (10% and 1.0%).
- In Checkmate 816, serious adverse reactions occurred in 30% of patients (n=176) who were treated with intravenous nivolumab in combination with platinum-doublet Serious adverse reactions in >2% included pneumonia and vomiting. No fatal adverse reactions occurred in patients who received intravenous nivolumab in combination with platinum-doublet chemotherapy. In Checkmate 77T, serious adverse reactions occurred in 21% of patients who received intravenous nivolumab in combination with platinum-doublet chemotherapy as neoadjuvant treatment (n=228). The most frequent (≥2%) serious adverse reactions was pneumonia. Fatal adverse reactions occurred in 2.2% of patients, due to cerebrovascular accident, COVID-19 infection, hemoptysis, pneumonia, and pneumonitis (0.4% each). In the adjuvant phase of Checkmate 77T, 22% of patients experienced serious adverse reactions (n=142). The most frequent serious adverse reaction was pneumonitis/ILD (2.8%). One fatal adverse reaction due to COVID-19 occurred. In Checkmate 017 and 057, serious adverse reactions occurred in 46% of patients receiving intravenous nivolumab (n=418). The most frequent serious adverse reactions reported in ≥2% of patients receiving intravenous nivolumab were pneumonia, pulmonary embolism, dyspnea, pyrexia, pleural effusion, pneumonitis, and respiratory failure. In Checkmate 057, fatal adverse reactions occurred; these included events of infection (7 patients, including one case of Pneumocystis jirovecii pneumonia), pulmonary embolism (4 patients), and limbic encephalitis (1 patient). In Checkmate 214, serious adverse reactions occurred in 59% of patients receiving intravenous nivolumab plus intravenous ipilimumab (n=547). The most frequent serious adverse reactions reported in ≥2% of patients were diarrhea, pyrexia, pneumonia, pneumonitis, hypophysitis, acute kidney injury, dyspnea, adrenal insufficiency, and colitis. In Checkmate 9ER, serious adverse reactions occurred in 48% of patients receiving intravenous nivolumab and cabozantinib (n=320). The most frequent serious adverse reactions reported in ≥2% of patients were diarrhea, pneumonia, pneumonitis, pulmonary embolism, urinary tract infection, and hyponatremia. Fatal intestinal perforations occurred in 3 (0.9%) patients.
- In Checkmate 025, serious adverse reactions occurred in 47% of patients receiving intravenous nivolumab (n=406). The most frequent serious adverse reactions reported in ≥2% of patients were acute kidney injury, pleural effusion, pneumonia, diarrhea, and In Checkmate 141, serious adverse reactions occurred in 49% of patients receiving intravenous nivolumab (n=236). The most frequent serious adverse reactions reported in ≥2% of patients receiving intravenous nivolumab were pneumonia, dyspnea, respiratory failure, respiratory tract infection, and sepsis. In Checkmate 275, serious adverse reactions occurred in 54% of patients receiving intravenous nivolumab (n=270). The most frequent serious adverse reactions reported in ≥ 2% of patients receiving intravenous nivolumab were urinary tract infection, sepsis, diarrhea, small intestine obstruction, and general physical health deterioration. In Checkmate 274, serious adverse reactions occurred in 30% of patients receiving intravenous nivolumab (n=351). The most frequent serious adverse reaction reported in ≥ 2% of patients receiving intravenous nivolumab was urinary tract infection. Fatal adverse reactions occurred in 1% of patients; these included events of pneumonitis (0.6%). In Checkmate 901, serious adverse reactions occurred in 48% of patients receiving intravenous nivolumab in combination with chemotherapy. The most frequent serious adverse reactions reported in ≥2% of patients who received intravenous nivolumab with chemotherapy were urinary tract infection (4.9%), acute kidney injury (4.3%), anemia (3%), pulmonary embolism (2.6%), sepsis (2.3%), and platelet count decreased (2.3%). Fatal adverse reactions occurred in 3.6% of patients who received intravenous nivolumab in combination with chemotherapy; these included sepsis (1%). In Checkmate 238, serious adverse reactions occurred in 18% of patients receiving intravenous nivolumab (n=452). Grade 3 or 4 adverse reactions occurred in 25% of intravenous nivolumab-treated patients (n=452). The most frequent Grade 3 and 4 adverse reactions reported in ≥2% of intravenous nivolumab-treated patients were diarrhea and increased lipase and amylase. In Attraction-3, serious adverse reactions occurred in 38% of patients receiving intravenous nivolumab (n=209). Serious adverse reactions reported in ≥2% of patients who received intravenous nivolumab were pneumonia, esophageal fistula, interstitial lung disease, and pyrexia. The following fatal adverse reactions occurred in patients who received intravenous nivolumab: interstitial lung disease or pneumonitis (1.4%), pneumonia (1.0%), septic shock (0.5%), esophageal fistula (0.5%), gastrointestinal hemorrhage (0.5%), pulmonary embolism (0.5%), and sudden death (0.5%). In Checkmate 577, serious adverse reactions occurred in 33% of patients receiving intravenous nivolumab (n=532). A serious adverse reaction reported in ≥2% of patients who received intravenous nivolumab was pneumonitis. A fatal reaction of myocardial infarction occurred in one patient who received intravenous nivolumab. In Checkmate 648, serious adverse reactions occurred in 62% of patients receiving intravenous nivolumab in combination with chemotherapy (n=310). The most frequent serious adverse reactions reported in ≥2% of patients who received intravenous nivolumab with chemotherapy were pneumonia (11%), dysphagia (7%), esophageal stenosis (2.9%), acute kidney injury (2.9%), and pyrexia (2.3%). Fatal adverse reactions occurred in 5 (1.6%) patients who received OPDIVO in combination with chemotherapy; these included pneumonitis, pneumatosis intestinalis, pneumonia, and acute kidney injury. In Checkmate 648, serious adverse reactions occurred in 69% of patients receiving intravenous nivolumab in combination with intravenous ipilimumab (n=322). The most frequent serious adverse reactions reported in ≥2% who received intravenous nivolumab in combination with intravenous ipilimumab were pneumonia (10%), pyrexia (4.3%), pneumonitis (4.0%), aspiration pneumonia (3.7%), dysphagia (3.7%), hepatic function abnormal (2.8%), decreased appetite (2.8%), adrenal insufficiency (2.5%), and dehydration (2.5%). Fatal adverse reactions occurred in 5 (1.6%) patients who received intravenous nivolumab in combination with intravenous ipilimumab; these included pneumonitis, interstitial lung disease, pulmonary embolism, and acute respiratory distress syndrome. In Checkmate 649, serious adverse reactions occurred in 52% of patients treated with intravenous nivolumab in combination with chemotherapy (n=782). The most frequent serious adverse reactions reported in ≥2% of patients treated with intravenous nivolumab in combination with chemotherapy were vomiting (3.7%), pneumonia (3.6%), anemia, (3.6%), pyrexia (2.8%), diarrhea (2.7%), febrile neutropenia (2.6%), and pneumonitis (2.4%). Fatal adverse reactions occurred in 16 (2.0%) patients who were treated with intravenous nivolumab in combination with chemotherapy; these included pneumonitis (4 patients), febrile neutropenia (2 patients), stroke (2 patients), gastrointestinal toxicity, intestinal mucositis, septic shock, pneumonia, infection, gastrointestinal bleeding, mesenteric vessel thrombosis, and disseminated intravascular coagulation. In Checkmate 76K, serious adverse reactions occurred in 18% of patients receiving intravenous nivolumab (n=524). Adverse reactions which resulted in permanent discontinuation of intravenous nivolumab in >1% of patients included arthralgia (1.7%), rash (1.7%), and diarrhea (1.1%). A fatal adverse reaction occurred in 1 (0.2%) patient (heart failure and acute kidney injury).
- The most frequent Grade 3-4 lab abnormalities reported in ≥1% of intravenous nivolumab-treated patients were increased lipase (2.9%), increased AST (2.2%), increased ALT (2.1%), lymphopenia (1.1%), and decreased potassium (1.0%).
Common Adverse Reactions
- In Checkmate 67T, the most common adverse reactions (≥10%) in patients treated with OPDIVO QVANTIG (n=247) were musculoskeletal pain (31%), fatigue (20%), pruritus (16%), rash (15%), hypothyroidism (12%), diarrhea (11%), cough (11%), and abdominal pain (10%). In Checkmate 037, the most common adverse reaction (≥20%) reported with intravenous nivolumab (n=268) was rash (21%). In Checkmate 066, the most common adverse reactions (≥20%) reported with intravenous nivolumab (n=206) vs dacarbazine (n=205) were fatigue (49% vs 39%), musculoskeletal pain (32% vs 25%), rash (28% vs 12%), and pruritus (23% vs 12%). In Checkmate 067, the most common (≥20%) adverse reactions in the intravenous nivolumab arm (n=313) were fatigue (59%), rash (40%), musculoskeletal pain (42%), diarrhea (36%), nausea (30%), cough (28%), pruritus (27%), upper respiratory tract infection (22%), decreased appetite (22%), headache (22%), constipation (21%), arthralgia (21%), and vomiting (20%). In Checkmate 067, the most common (≥20%) adverse reactions in the intravenous nivolumab plus intravenous ipilimumab arm (n=313) were fatigue (62%), diarrhea (54%), rash (53%), nausea (44%), pyrexia (40%), pruritus (39%), musculoskeletal pain (32%), vomiting (31%), decreased appetite (29%), cough (27%), headache (26%), dyspnea (24%), upper respiratory tract infection (23%), arthralgia (21%), and increased transaminases (25%).
- In Checkmate 816, the most common (>20%) adverse reactions in the intravenous nivolumab plus chemotherapy arm (n=176) were nausea (38%), constipation (34%), fatigue (26%), decreased appetite (20%), and rash (20%). In Checkmate 77T, the most common adverse reactions (reported in ≥20%) in patients receiving intravenous nivolumab in combination with chemotherapy (n= 228) were anemia (39.5%), constipation (32.0%), nausea (28.9%), fatigue (28.1%), alopecia (25.9%), and cough (21.9%). In Checkmate 017 and 057, the most common adverse reactions (≥20%) in patients receiving intravenous nivolumab (n=418) were fatigue, musculoskeletal pain, cough, dyspnea, and decreased appetite. In Checkmate 214, the most common adverse reactions (≥20%) reported in patients treated with intravenous nivolumab plus intravenous ipilimumab (n=547) were fatigue (58%), rash (39%), diarrhea (38%), musculoskeletal pain (37%), pruritus (33%), nausea (30%), cough (28%), pyrexia (25%), arthralgia (23%), decreased appetite (21%), dyspnea (20%), and vomiting (20%). In Checkmate 9ER, the most common adverse reactions (≥20%) in patients receiving intravenous nivolumab and cabozantinib (n=320) were diarrhea (64%), fatigue (51%), hepatotoxicity (44%), palmar-plantar erythrodysaesthesia syndrome (40%), stomatitis (37%), rash (36%), hypertension (36%), hypothyroidism (34%), musculoskeletal pain (33%), decreased appetite (28%), nausea (27%), dysgeusia (24%), abdominal pain (22%), cough (20%) and upper respiratory tract infection (20%). In Checkmate 025, the most common adverse reactions (≥20%) reported in patients receiving intravenous nivolumab (n=406) vs everolimus (n=397) were fatigue (56% vs 57%), cough (34% vs 38%), nausea (28% vs 29%), rash (28% vs 36%), dyspnea (27% vs 31%), diarrhea (25% vs 32%), constipation (23% vs 18%), decreased appetite (23% vs 30%), back pain (21% vs 16%), and arthralgia (20% vs 14%). In Checkmate 141, the most common adverse reactions (≥10%) in patients receiving intravenous nivolumab (n=236) were cough (14%) and dyspnea (14%) at a higher incidence than investigator’s In Checkmate 275, the most common adverse reactions (≥ 20%) reported in patients receiving intravenous nivolumab (n=270) were fatigue (46%), musculoskeletal pain (30%), nausea (22%), and decreased appetite (22%). In Checkmate 274, the most common adverse reactions (20%) reported in patients receiving intravenous nivolumab (n=351) were rash (36%), fatigue (36%), diarrhea (30%), pruritus (30%), musculoskeletal pain (28%), and urinary tract infection (22%). In Checkmate 901, the most common adverse reactions (reported in ≥20% of patients) were nausea (52%), fatigue (48%), musculoskeletal pain (33%), constipation (30%), decreased appetite (30%), rash (25%), vomiting (23%), and peripheral neuropathy (20%). In Checkmate 238, the most common adverse reactions (≥20%) reported in intravenous nivolumab-treated patients (n=452) vs ipilimumab-treated patients (n=453) were fatigue (57% vs 55%), diarrhea (37% vs 55%), rash (35% vs 47%), musculoskeletal pain (32% vs 27%), pruritus (28% vs 37%), headache (23% vs 31%), nausea (23% vs 28%), upper respiratory infection (22% vs 15%), and abdominal pain (21% vs 23%). The most common immune-mediated adverse reactions were rash (16%), diarrhea/colitis (6%), and hepatitis (3%). In Attraction-3, the most common adverse reactions (≥20%) in intravenous nivolumab-treated patients (n=209) were rash (22%) and decreased appetite (21%). In Checkmate 577, the most common adverse reactions (≥20%) in patients receiving intravenous nivolumab (n=532) were fatigue (34%), diarrhea (29%), nausea (23%), rash (21%), musculoskeletal pain (21%), and cough (20%). In Checkmate 648, the most common adverse reactions (≥20%) in patients treated with intravenous nivolumab in combination with chemotherapy (n=310) were nausea (65%), decreased appetite (51%), fatigue (47%), constipation (44%), stomatitis (44%), diarrhea (29%), and vomiting (23%). In Checkmate 648, the most common adverse reactions reported in ≥20% of patients treated with intravenous nivolumab in combination with intravenous ipilimumab were rash (31%), fatigue (28 %), pyrexia (23%), nausea (22%), diarrhea (22%), and constipation (20%). In Checkmate 649, the most common adverse reactions (≥20%) in patients treated with intravenous nivolumab in combination with chemotherapy (n=782) were peripheral neuropathy (53%), nausea (48%), fatigue (44%), diarrhea (39%), vomiting (31%), decreased appetite (29%), abdominal pain (27%), constipation (25%), and musculoskeletal pain (20%). In Checkmate 76K, the most common adverse reactions (≥20%) reported with intravenous nivolumab (n=524) were fatigue (36%), musculoskeletal pain (30%), rash (28%), diarrhea (23%) and pruritus (20%).
Surgery Related Adverse Reactions
- In Checkmate 77T, 5.3% (n=12) of the intravenous nivolumab-treated patients who received neoadjuvant treatment, did not receive surgery due to adverse reactions. The adverse reactions that led to cancellation of surgery in intravenous nivolumab-treated patients were cerebrovascular accident, pneumonia, and colitis/diarrhea (2 patients each) and acute coronary syndrome, myocarditis, hemoptysis, pneumonitis, COVID-19, and myositis (1 patient each).
Please see US Full Prescribing Information for OPDIVO QVANTIG.
References:
- OPDIVO Qvantig [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
- OPDIVO [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
- Bittner B et al. BioDrugs. 2018;32:425-440.
- Albiges L et al. Ann Oncol. 2025;36(1):99-107.
- Bittner B, Schmidt J. BioDrugs. 2024;38(1):23-46.
- George S et al. Oral presentation at ASCO GU 2024. Abstract LBA360.
© 2025 Bristol-Myers Squibb Company. OPDIVO QvantigTM and the related logo are trademarks of Bristol-Myers Squibb Company.
1992-US-2500197 05/25
