
In a large International Premenopausal Breast Cancer study which included close to 900,000 women, compared with nulliparous women, parous women had an increased risk for breast cancer that peaked about 5 years after childbirth and then gradually decreased about 24 years after childbirth.The increase in breast cancer risk after childbirth may be due to proliferation of breast cells during pregnancy which could promote accelerated development of latent initiated tumor cells. Childbirth also brings about maternal changes beyond breast tissue including altered immune function and microbiota, increased stress, and accelerated aging processes. Health Care Professionals should take these factors into account when considering individual risk profiles for breast cancer in premenopausal women.
Tag: Breast Cancer
Increased Risk of Breast Cancer after Recent Childbirth
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 268,600 new cases of female breast cancer will be diagnosed in 2019 and about 41,760 women will die of the disease. Breast cancer is the most common cancer type in reproductive-aged women. Women with biological children (parous women) are at a lower risk for developing breast cancer compared to nulliparous women. However, parity as a protective factor largely applies to breast cancer developing after age 60 years and may not apply for younger premenopausal women. Evidence from national registry linkage studies in Scandinavian countries suggested that recent childbirth confers a short-term increase in breast cancer risk which may last for 10 years or more, and this risk may be further increased in women who are older at first childbirth. Studies published thus far have not shown consistent findings and have had limited ability to account for factors influencing breast cancer risk such as breastfeeding and family history of breast cancer.
One biological explanation for an increase in breast cancer risk after childbirth may be due to proliferation of breast cells during pregnancy which could promote accelerated development of latent initiated tumor cells. This may also explain the higher breast cancer risk, conferred by older age at first childbirth, as a result of higher proportion of latent initiated tumor cells at older ages. Further, the postpartum breast microenvironment, characterized by lactational involution may also facilitate cancer cell migration and metastasis. Childbirth also brings about maternal changes beyond breast tissue including altered immune function and microbiota, increased stress, and accelerated aging processes.
The authors in this study used data from the International Premenopausal Breast Cancer Collaborative Group and conducted a pooled analysis of individual-level data from 15 prospective cohort studies. In this analysis, the researchers included women younger than 55 years and evaluated the risk of breast cancer in relation to recent childbirth, while taking into account other factors that relate to breast cancer risk such as breastfeeding, numbers of pregnancies and births and family history of breast cancer. It is felt that understanding these risk patterns may have implications for identifying risk-reducing strategies among vulnerable subgroups. A total of 889, 944 women, were available for analysis after excluding women who reported a first birth before age 13 years, women who were 50 years or older at study entry and at most recent birth, or reached parity greater than 10 births before enrollment. All these events were considered to have greater potential for data errors. The mean age at study entry was 42 years.
The researchers noted that compared with nulliparous women, parous women had an increased risk for breast cancer that peaked about 5 years after childbirth and then gradually decreased about 24 years after childbirth. These findings however were not noted among women who had only 1 child, or had their first child before age 25 years. The risk was highest in women who were older at the time of first childbirth, multiparous women and those who had a family history of breast cancer. Among those women with a family history of breast cancer, the risk was the greatest for Estrogen Receptor negative breast cancer. Breast feeding did not influence breast cancer risk patterns.
It was concluded that compared with nulliparous women, parous women have an increased risk for breast cancer after childbirth that is highest the first 5 years but decreases over the following 20 years. Health Care Professionals should take these factors into account when considering individual risk profiles for breast cancer in premenopausal women. Breast Cancer Risk After Recent Childbirth: A Pooled Analysis of 15 Prospective Studies. Nichols HB, Schoemaker MJ, Cai J, et al. Ann Intern Med. 2019;170:22-30
ONTRUZANT® (Trastuzumab-dttb)
The FDA on January 18, 2019 granted approval to ONTRUZANT®, a Trastuzumab (HERCEPTIN®) biosimilar, for the treatment of patients with HER2-overexpressing Breast cancer or metastatic Gastric or GastroEsophageal Junction adenocarcinoma. The newly approved biosimilar will also eventually compete with 2 prior FDA approved biosimilars, OGIVRI® (Trastuzumab-dkst) and HERZUMA® (Trastuzumab-pkrb). ONTRUZANT® is a product of Samsung Bioepis and will be marketed by Merck&Co.
Late Breaking Abstract – ESMO 2018 Targeting PIK3CA Mutations Improves Outcomes in Advanced Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 268,600 new cases of female breast cancer will be diagnosed in 2019 and about 41,760 women will die of the disease. About 70% of breast tumors express Estrogen Receptors and/or Progesterone Receptors and these patients are often treated with anti-estrogen therapy as first line treatment. However, resistance to hormonal therapy occurs in a majority of the patients.
The PhosphoInositide 3-Kinase (PI3K) pathway is an intracellular signaling pathway important in the regulation of cancer cell proliferation and metastasis. PI3K is a lipid kinase and has four distinct isoforms – alpha, beta, gamma and delta, which play a unique role in the survival of different tumor types and establishment of supportive tumor microenvironments. The alpha and beta isoforms are expressed in a wide variety of tissues whereas the gamma and delta isoforms are primarily expressed in hematopoietic cells such as B and T cells. The PI3K alpha isoform is particularly important in breast cancer and plays an important role in tumorigenesis, supporting tumor angiogenesis and stromal interactions, making this a viable target. PIK3CA is an oncogene that codes for the alpha isoform of PI3K, (PI3Kα), more specifically for the alpha isoform of p110. The PI3k pathway is the most frequently altered pathway in human cancers including breast cancer, and has been implicated in disease progression in a significant number of patients with breast cancer. Activation of the PI3K pathway in breast cancer has been associated with resistance to endocrine therapy and disease progression. Approximately 40% of patients with Hormone Receptor positive (HR+), HER2 negative breast cancers, harbor activating mutations in the PIK3CA isoform of PI3K, which is the most common mutation in HR+ breast cancer. Patients with advanced breast cancer harboring PIK3CA mutations typically have a poor prognosis. This provides a strong rationale for targeting the PI3K pathway in breast cancer.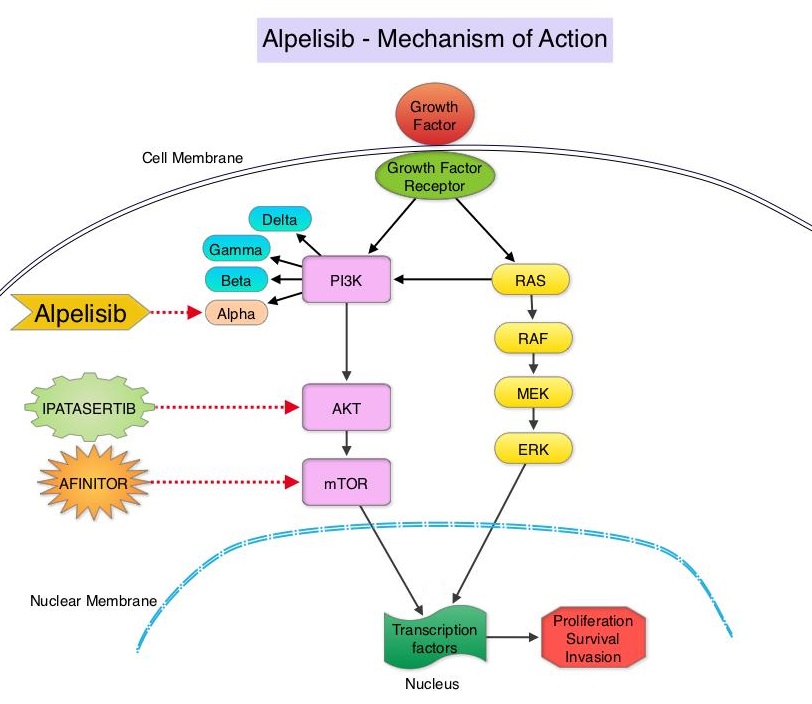
Alpelisib is an oral, alpha-specific PI3K inhibitor that specifically inhibits PIK3 in the PI3K/AKT kinase signaling pathway. Further, it was shown in preclinical studies that cancer cells with PIK3CA mutations are more sensitive to Alpelisib than those without the mutation, across a broad range of tumor types. SOLAR-1 clinical trial was conducted to test this hypothesis.
SOLAR-1 is a global, double-blind, placebo-controlled, randomized phase III trial, which studied the benefit of Alpelisib in combination with FASLODEX® (Fulvestrant) among postmenopausal women and men with PIK3CA-mutated HR+/HER2 negative advanced or metastatic breast cancer, who had progressed on or following prior Aromatase Inhibitor treatment with or without a Cyclin-Dependent Kinase (CDK) 4/6 inhibitor. In this study, 572 patients were randomized in a 1:1 ratio to receive Alpelisib 300 mg orally daily or placebo once daily, in combination with FASLODEX® 500mg IM on days 1 and 15 of the first cycle and day 1 of each subsequent 28-day cycle. Patients were stratified based on visceral metastases and prior CDK4/6 inhibitor treatment. A total of 341 patients had PIK3CA mutations upon testing of the tumor tissue with 169 patients receiving the Alpelisib combination and 172 patients receiving FASLODEX® alone. Enrolled patients had received one or more prior lines of hormonal therapy, but no chemotherapy for advanced breast cancer. They had not previously received FASLODEX® or any PI3K, Akt or mTOR inhibitor, and were not on concurrent anticancer therapy. Approximately half of the patients in each treatment group had lung or liver metastases and 6% had received prior CDK4/6 inhibitor therapy. The Primary endpoint was Progression Free Survival (PFS) for patients with the PIK3CA mutation. Secondary endpoints included Overall Survival (OS), Overall Response Rate (ORR), Clinical Benefit Rate, Health-Related Quality of Life, Efficacy in PIK3CA non-mutant cohort, Safety and Tolerability.
The Primary endpoint was met and at a median follow up of 20 months, the PFS was nearly twice as long in patients with PIK3CA mutations randomized to Alpelisib plus FASLODEX® compared to the placebo plus FASLODEX® group. The median PFS was 11.0 months in the Alpelisib group compared to 5.7 months in the placebo group (HR=0.65; P=0.00065). In patients with measurable, PIK3CA-mutated advanced breast cancer (N=262), the Overall Response Rate was 36% for the Alpelisib plus FASLODEX® group versus 16% for placebo plus FASLODEX® group (P=0.0002). There was no significant PFS benefit noted in the PIK3CA-nonmutant patient group receiving Alpelisib plus FASLODEX®. The most frequent toxicities with Alpelisib were hyperglycemia which could be managed with Metformin, nausea, decreased appetite and rash.
It was concluded that Alpelisib given along with FASLODEX® significantly improved Progression Free Survival compared to Placebo plus FASLODEX®, with manageable toxicities. The authors commented that this is the first study to show statistically significant, clinically meaningful PFS improvement with an alpha-specific PI3K inhibitor in PIK3CA-mutated HR+, HER2 negative advanced breast cancer, highlighting the importance of clinical genomics in advanced breast cancer. It however remains unclear whether Alpelisib should be incorporated into the current treatment paradigm upfront, along with endocrine therapy and a CDK 4/6 inhibitor, or sequentially following disease progression on a combination of endocrine therapy and a CDK 4/6 inhibitor. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the phase 3 SOLAR-1 trial. André F, Ciruelos EM, Rubovszky G, et al. Presented during the Presidential Symposium 1 at: 2018 ESMO Congress; October 19-23; Munich, Germany. Abstract LBA3_PR.
HERZUMA® (Trastuzumab-pkrb)
The FDA on December 14, 2018 approved HERZUMA® as a biosimilar to HERCEPTIN® (Trastuzumab) for patients with HER2-overexpressing breast cancer. HERZUMA® is a product of Celltrion Inc.
Axillary Radiotherapy is an Alternative to Complete Lymph Node Dissection in Early-Stage Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 266,120 new cases of invasive breast cancer will be diagnosed in 2018 and about 40,920 women will die of the disease. Axillary lymph node evaluation is an important part of breast cancer staging and the presence of axillary lymph metastases decreases the 5-year survival rate by 28-40%. Axillary lymph node status remains the most powerful predictor of breast cancer recurrence and survival. Axillary Lymph Node Dissection (ALND) was first advocated in the 18th century as part of the treatment of invasive breast cancer and has been standard practice until 2 decades back. ALND can be associated with significant morbidities such as upper limb lymphedema, pain, and sensitivity disorders and this can have a major psychological impact on breast cancer patients. Sentinel Lymph Node Biopsy (SLNB) which was introduced into clinical practice in the mid 1990’s, however has now become the standard for Stage I and II breast cancer. The sentinel node is the first lymph node(s) to which cancer cells are most likely to metastasize from a primary tumor. With the introduction of intraoperative lymphatic mapping in the 1990s, Sentinel Lymph Node Biopsy (SLNB) has now gained general acceptance and if the sentinel node is negative for metastatic disease or only has minimal disease, then no further axillary surgery is indicated. Unlike Axillary Lymph Node Dissection (ALND), SLNB is associated with a lower incidence of lymphedema/ seroma at the surgery site, paresthesias and restriction of joint movement. Nine randomized clinical trials have not shown any difference in mortality among patients who underwent ALND or SLNB for either lymph node metastases or negative sentinel lymph nodes, validating Sentinel Lymph Node Biopsy (SLNB). The American Society of Clinical Oncology (ASCO) first published guidelines on the use of SLNB for patients with early stage breast cancer in 2005, based on one randomized clinical trial. Since then, ASCO updated Clinical Practice Guideline based on additional information from 9 randomized clinical trials and13 cohort studies pertinent to SLNB and ALND.
Patients with T1-2 tumors with positive Sentinel Lymph Node Biopsy usually undergo complete ALND and there is increasing controversy about whether ALND is always necessary. AMAROS is a multicenter, randomized phase III trial, sponsored by the European Organisation for Research and Treatment of Cancer (EORTC), in which the effectiveness of complete Axillary Lymph Node Dissection (ALND) was compared with axillary Radiation Therapy (RT), in patients with invasive breast cancer. The rationale was that Radiation Therapy uses high-energy x-rays to damage tumor cells and may be a less invasive treatment and causes fewer side effects than complete ALND. This study was conducted to evaluate whether axillary RT could yield comparable outcomes to ALND with fewer adverse side effects, in this patient population. This trial enrolled 4806 patients with early-stage, clinically node-negative breast cancer of whom 1425 patients had a positive sentinel lymph node biopsy. Of these patients, 744 were randomly assigned to undergo complete ALND, whereas 681 patients received axillary RT. Both treatment groups were well balanced. The first 5-year follow up data published in 2013 showed that upper extremity lymphedema occurred significantly less often in those who received Radiotherapy compared with those who underwent complete ALND, and recent Quality of life and morbidity data supported these earlier findings.
The authors herein presented the 10 year follow up data of the AMAROS trial. It was noted that at 10 years, 1.82% of patients assigned to axillary RT had an axillary recurrence compared with 0.93% of those assigned to complete ALND, and this suggested that there was no significant difference (P=0.36). Further, there was no significant difference in the distant metastasis–free survival or Overall Survival between the two treatment groups. The distant metastasis–free survival was 78.2% among those assigned to axillary RT and 81.7% among those assigned to complete ALND and Overall Survival in the two treatment groups was 81.4% and 84.6%, respectively. It was noted that there was a higher 10-year cumulative incidence of second primaries among patients assigned to axillary RT compared with those assigned to complete Axillary Lymph Node Dissection (12% versus 8.3%). It remains unclear whether the addition of the radiation will increase the risk of second primary cancers.
It was concluded that axillary Radiotherapy is noninferior to complete Axillary Lymph Node Dissection in terms of locoregional control and this trial suggests that some patients with a positive sentinel lymph node biopsy may be appropriate candidates for axillary Radiotherapy. Rutgers, E. Radiotherapy or surgery of the axilla after a positive sentinel node in breast cancer patients: 10-year results of the EORTC AMAROS trial. Presented at the 2018 San Antonio Breast Cancer Symposium; December 4-8; San Antonio, Texas.(Abstract GS4-01)
Adjuvant KADCYLA® Superior to HERCEPTIN® in High Risk HER2-Positive Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 266,120 new cases of invasive breast cancer will be diagnosed in 2018 and about 40,920 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. HERCEPTIN® (Trastuzumab) is a humanized monoclonal antibody targeting HER2, and adjuvant and neoadjuvant chemotherapy given along with HERCEPTIN® reduces the risk of disease recurrence and death, among patients with HER2-positive, early stage as well as advanced metastatic breast cancer. Since the approval of HERCEPTIN®, several other HER2-targeted therapies have become available. The duration of adjuvant HERCEPTIN® therapy has been 12 months and this length of treatment was empirically adopted from the pivotal registration trials.
KADCYLA® (Ado-Trastuzumab Emtansine, T-DM1) is an Antibody-Drug Conjugate (ADC) comprised of the antibody HERCEPTIN® and the chemotherapy agent Emtansine, linked together. Upon binding to the HER2 receptor, it not only inhibits the HER2 signaling pathways but also delivers a chemotherapy agent Emtansine, a microtubule inhibitor, directly inside the tumor cells. This agent is internalized by lysosomes and destroys the HER2-positive tumor cells upon intracellular release. In the EMILIA trial, KADCYLA® was associated with significant increase in Overall Survival when compared with TYKERB® (Lapatinib) plus XELODA® (Capecitabine), in HER2-positive metastatic breast cancer patients, who had previously received HERCEPTIN® and a Taxane.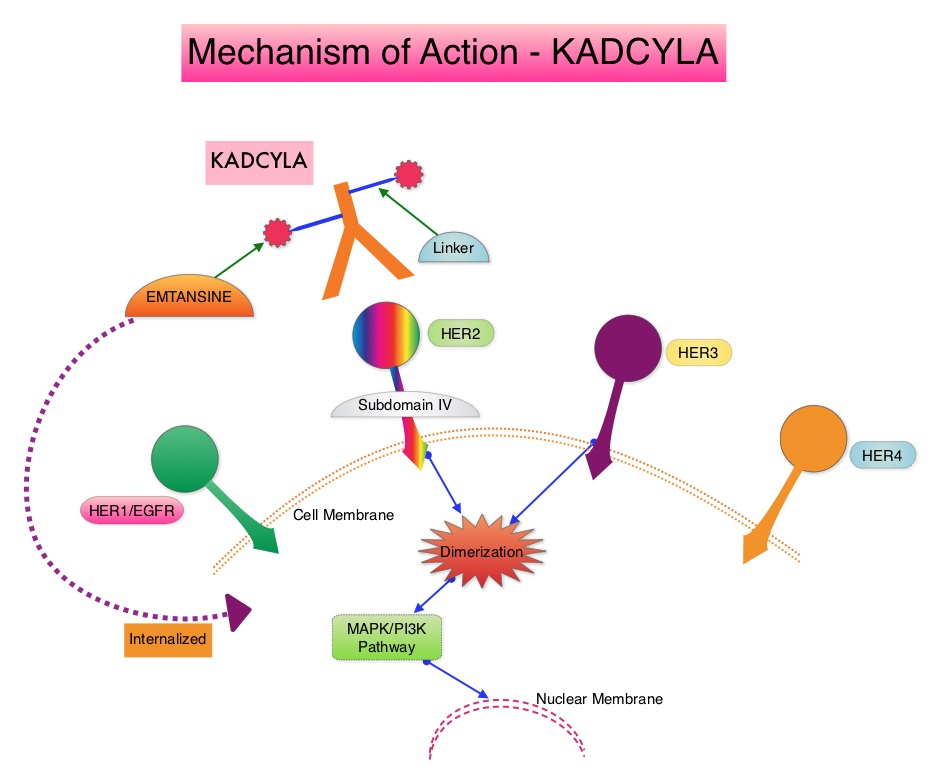
The present study was conducted to address the unmet need of patients who have residual invasive breast cancer after receiving neoadjuvant chemotherapy plus HER2-targeted therapy. These high risk patients have an unfavorable prognosis, compared to those who have no residual cancer following neoadjuvant therapy. The KATHERINE trial is an open-label, phase III global study, which compared KADCYLA® with HERCEPTIN®, as an adjuvant treatment for patients with HER2-positive early breast cancer, who had residual invasive disease following neoadjuvant chemotherapy and HERCEPTIN®.
This study included 1,486 patients with HER2-positive early stage breast cancer, who were found to have residual invasive disease in the breast or axillary lymph nodes at surgery, following at least six cycles (16 weeks) of neoadjuvant chemotherapy with a Taxane (with or without Anthracycline) and HERCEPTIN®. Within 12 weeks of surgery, patients (N=1486) were randomly assigned in a 1:1 ratio to KADCYLA® 3.6 mg/kg IV every 3 weeks or HERCEPTIN® 6 mg/kg IV every 3 weeks, for 14 cycles (743 patients in each group). Both treatment groups were well balanced and hormone receptor positive disease was present in 72% of the patients. The majority of the patients (77%) had received an Anthracycline-containing neoadjuvant chemotherapy regimen, and in 19% of the patients, another HER2-targeted agent in addition to HERCEPTIN® had been administered as a component of neoadjuvant therapy. The Primary end point was invasive Disease Free Survival (defined as freedom from ipsilateral invasive breast tumor recurrence, ipsilateral locoregional invasive breast cancer recurrence, contralateral invasive breast cancer, distant recurrence, or death from any cause). The median duration of follow up was 41.4 months in the KADCYLA® group and 40.9 months in the HERCEPTIN® group.
At the prespecified interim analysis, invasive disease occurred in 12.2% of patients who received KADCYLA® and 22.2% of patients who received HERCEPTIN®. The estimated percentage of patients who were free of invasive disease at 3 years was 88.3% in the KADCYLA® group and 77.0% in the HERCEPTIN® group. Invasive Disease Free Survival which was the Primary end point of the study was significantly higher in the KADCYLA® group than in the HERCEPTIN® group (HR=0.50; P<0.001). This suggested that KADCYLA® reduced the risk of developing an invasive breast cancer recurrence or death by 50%. Distant recurrence as the first invasive disease event occurred in 10.5% of patients in the KADCYLA® group and in 15.9% of the HERCEPTIN® group. A consistent benefit was seen across all prespecified subgroups. Adverse events were consistent with the known safety profile of KADCYLA®, with more toxicities associated with KADCYLA® than with HERCEPTIN®. Additional follow-up will be necessary to determine the Overall Survival benefit with adjuvant KADCYLA®.
It was concluded that among patients with HER2-positive early breast cancer who had residual invasive disease after completion of neoadjuvant therapy, substituting KADCYLA® for adjuvant HERCEPTIN® reduced the risk of recurrence of invasive breast cancer or death was 50%, with the benefit seen across all patient subgroups. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. von Minckwitz G, Huang C-S, Mano MS, et al. for the KATHERINE Investigators. (published online December 5, 2018). New Engl Jour Med. doi: 10.1056/NEJMoa1814017
TALZENNA® (Talazoparib)
The FDA on October 16, 2018 approved TALZENNA®, a poly (ADP-ribose) polymerase (PARP) inhibitor, for patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm), HER2 negative, locally advanced or metastatic breast cancer. Patients must be selected for therapy based on an FDA-approved companion diagnostic for TALZENNA®. TALZENNA® is a product of Pfizer Inc.
Testing for BRCA1 and BRCA2 Mutations May Not Be Adequate in Breast Cancer Patients
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. It is estimated that 266,120 new cases of invasive breast cancer will be diagnosed in 2018 and 40,920 women are expected to die from the disease. DNA can be damaged due to errors during its replication or as a result of environmental exposure to ultraviolet radiation from the sun or other toxins. The tumor suppressor genes such as BRCA1 (Breast Cancer 1) and BRCA2 help repair damaged DNA and thus play an important role in maintaining cellular genetic integrity, failing which these genetic aberrations can result in malignancies. The BRCA1 gene is located on the long (q) arm of chromosome 17 whereas BRCA2 is located on the long arm of chromosome 13. Mutations in BRCA1 and BRCA2 account for about 20 to 25 percent of hereditary breast cancers and about 5 to 10 percent of all breast cancers.These mutations can be inherited from either of the parents and a child has a 50 percent chance of inheriting this mutation and the deleterious effects of the mutations are seen even when an individual’s second copy of the gene is normal.
Breast cancer patients have a 5-12% lifetime risk of a second primary cancer. It remains unclear however whether patients with breast cancer and another primary cancer have mutations in genes other than BRCA1 and BRCA2, compared to those with a single breast cancer. There are well established data on the cancer risks, associated with different gene mutations. The authors hypothesized that among these patients, a number of factors including environmental exposures and genetic predisposition, may play a role in the development of more than one primary cancer in their lifetime. Recently published study suggested that there was a 85% cumulative breast cancer risk by age 60 years, among those with mutations in the TP53 gene (Cancer2016;122(23):3673-3681). Further with the increasing recognition that germline mutations in genes may have clinical and treatment implications, majority of patients are feeling comfortable opting for upfront multiple genetic mutation testing.
The researchers in this study looked at a panel of 15 actionable mutations beyond BRCA and the gene panel included TP53, PALB2, CDH1, PTEN, STK11, CHEK2, ATM, NBN, MSH6, PMS2, MSH2, MLH1, CDKN2A, MUTYH monoallelic, and CHEK2 Low Risk. Two cohorts of BRCA1 and BRCA2 negative patients were studied. The first cohort included high-risk breast cancer patients with either a single breast cancer (N=464) or breast cancer and an additional primary cancer (N=551). The second cohort comprised of patients with familial breast cancer (inherited risk) with either a single breast cancer (N=1464) or breast cancer and another primary cancer (N=340).
In a total of 891 patients in both cohorts who had breast cancer and an additional primary cancer, there was twice the risk of inheriting mutations in genes other than BRCA1 and BRCA2. In cohort 1, the mutation rate among patients who had breast cancer and an additional primary cancer was 8.7% compared to 4.1% among those with single breast cancer (P=0.003) and in cohort 2, the mutation rate was 8.2% versus 4.2%, respectively (P=0.003).
There was however a differences in individual gene mutation rates between the two cohorts. Among patients with breast cancer and an additional primary cancer in cohort 1, mutations in TP53 and MSH6 were significantly higher, whereas among the patients in cohort 2 with familial breast cancer, mutations in ATM, CHEK2 and PALB2 were significantly higher both in those with breast cancer and another primary cancer and those with a single breast cancer.
The authors concluded that patients with multiple primary cancers should be offered multiplex panel testing to identify patients at risk. Identifying mutations, especially mutations in the TP53 gene may have a bearing on appropriate recommendations such as risk-reducing bilateral mastectomy or mastectomy instead of a lumpectomy in this patient group. Thus, risk assessment using multiple genetic testing panels can be beneficial for clinical care and surveillance. Inherited mutations in breast cancer patients with and without multiple primary cancers. Maxwell KN, Vijai V, Lilyquist J, et al. DOI: 10.1200/JCO.2018.36.15_suppl.1503 Journal of Clinical Oncology 36, no. 15_suppl (May 2018) 1503-1503.
ASCO Endorses Complementary Therapy Guidelines for Breast Cancer Patients
SUMMARY: The American Society for Clinical Oncology (ASCO) after careful review, endorsed the Society for Integrative Oncology (SIO) guideline on the use of integrative therapies during and after breast cancer treatment. Integrative medicine is defined as a patient-centered, evidence-informed field of care that uses mind and body practices, natural products, and/or lifestyle modifications to improve health, quality of life, and clinical outcomes. The ASCO Expert Panel determined that the recommendations in the SIO guideline published in 2017 are clear, thorough, and based on the most relevant scientific evidence. The panel emphasized that these therapies are complementary and should be used along with conventional anticancer treatment, thereby taking a more comprehensive treatment approach across the cancer care continuum, to manage symptoms and adverse effects of breast cancer treatment. This ASCO guideline excluded lifestyle changes such as diet and exercise, mainstream interventions such as support groups, and practices like attention-restoration therapy, that are still being evaluated. Practices such as prayer and spirituality were not considered specifically integrative oncology therapy.
Guideline Question
What are evidence-based approaches to the use of integrative therapies in the management of symptoms and adverse effects during and after breast cancer treatment?
Target Population
Patients undergoing treatment of breast cancer and survivors of breast cancer
Target Audience
Oncologists, integrative medicine providers, supportive care specialists, nurses, pharmacists, primary care providers, and patients with breast cancer

Key Recommendations
Acute Radiation Skin Reaction
Aloe vera and hyaluronic acid cream should not be recommended for improving acute radiation skin reaction. (Grade D)
Anxiety and Stress Reduction
Meditation is recommended for reducing anxiety. (Grade A)
Music therapy is recommended for reducing anxiety. (Grade B)
Stress management is recommended for reducing anxiety during treatment, but longer group programs are likely better than self-administered home programs or shorter programs. (Grade B)
Yoga is recommended for reducing anxiety. (Grade B)
Acupuncture, massage, and relaxation can be considered for reducing anxiety. (Grade C)
Chemotherapy-Induced Nausea and Vomiting
Acupressure can be considered as an addition to antiemetic drugs to control nausea and vomiting during chemotherapy. (Grade B)
Electroacupuncture can be considered as an addition to antiemetic drugs to control vomiting during chemotherapy. (Grade B)
Ginger and relaxation can be considered as additions to antiemetic drugs to control nausea and vomiting during chemotherapy. (Grade C)
Glutamine should not be recommended for improving nausea and vomiting during chemotherapy. (Grade D)
Depression and Mood Disturbance
Meditation, particularly mindfulness-based stress reduction, is recommended for treating mood disturbance and depressive symptoms. (Grade A)
Relaxation is recommended for improving mood disturbance and depressive symptoms. (Grade A)
Yoga is recommended for improving mood disturbance and depressive symptoms. (Grade B)
Massage is recommended for improving mood disturbance. (Grade B)
Music therapy is recommended for improving mood disturbance. (Grade B)
Acupuncture, healing touch, and stress management can be considered for improving mood disturbance and depressive symptoms. (Grade C)
Fatigue
Hypnosis and ginseng can be considered for improving fatigue during treatment. (Grade C)
Acupuncture and yoga can be considered for improving post-treatment fatigue. (Grade C)
Acetyl-l-carnitine and guarana should not be recommended for improving fatigue during treatment. (Grade D)
Lymphedema
Low-level laser therapy, manual lymphatic drainage, and compression bandaging can be considered for improving lymphedema. (Grade C)
Neuropathy
Acetyl-l-carnitine is not recommended for the prevention of chemotherapy-induced peripheral neuropathy in patients with breast cancer due to potential harm. (Grade H)
Pain
Acupuncture, healing touch, hypnosis, and music therapy can be considered for the management of pain. (Grade C)
Quality of Life
Meditation is recommended for improving quality of life. (Grade A)
Yoga is recommended for improving quality of life. (Grade B)
Acupuncture, mistletoe, qigong, reflexology, and stress management can be considered for improving quality of life. (Grade C)
Sleep Disturbance
Gentle yoga can be considered for improving sleep. (Grade C)
Vasomotor/Hot Flashes
Acupuncture can be considered for improving hot flashes. (Grade C)
Soy is not recommended for hot flashes in patients with breast cancer due to lack of effect. (Grade D)
Integrative Therapies During and After Breast Cancer Treatment: ASCO Endorsement of the SIO Clinical Practice Guideline. Lyman GH, Greenlee H, Bohlke K, et al. J Clin Oncol. 2018;36:2647-2655