
SUMMARY: The FDA on April 28, 2017 granted accelerated approval to ALUNBRIG® (Brigatinib),for the treatment of patients with metastatic Anaplastic Lymphoma Kinase (ALK)-positive Non Small Cell Lung Cancer (NSCLC), who have progressed on or are intolerant to XALKORI® (Crizotinib). Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 222,500 new cases of lung cancer will be diagnosed in the United States in 2017 and over 155,870 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 25% are Squamous Cell Carcinomas, 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. The discovery of rearrangements of the Anaplastic Lymphoma Kinase (ALK) gene in some patients with advanced NSCLC and adenocarcinoma histology, led to the development of agents such as XALKORI® (Crizotinib), ZYKADIA® (Ceritinib), ALECENSA® (Alectinib) and now ALUNBRIG® (Brigatinib), with promising results. It has become clear that appropriate, molecularly targeted therapy for tumors with a molecular abnormality, results in the best outcomes. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8% as well as other mutations in BRAF, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive, and the presence of two simultaneous mutations are rare.
The approval was based on findings from the ALTA trial, which is a pivotal, open-label multicenter study in which 222 patients were randomized 1:1 ratio to receive ALUNBRIG® 90 mg orally once daily (N=112) or 180 mg once daily following a 7-day lead-in at 90 mg orally once daily (N=110). Two dose regimens were evaluated in this study, as clinical responses and Adverse Events varied with starting dose of ALUNBRIG®, in a previous Phase I/II study. Eligible patients had locally advanced or metastatic ALK-positive NSCLC, who had progressed on XALKORI®. The median age of patients across the study was 54 years and 67% of the patients had brain metastases. Patients were stratified by presence of brain metastases at baseline and best response to prior treatment with XALKORI®. The Primary endpoint was Objective Response Rate (ORR) and Secondary endpoints included Progression Free Survival (PFS) and CNS response.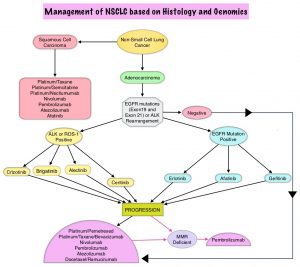
The ORR was 48% in the 90 mg dose group and 53% in the 180 mg dose group. After a median duration of follow up of 8 months, median duration of response was 13.8 months in both treatment groups. In patients with measurable brain metastases at baseline, intracranial ORR was 42% in the 90 mg group and 67% in the 180 mg group. Among patients who exhibited an intracranial response, 78% of patients in the 90 mg group and 68% of patients in the 180 mg group maintained an intracranial response for at least 4 months. The median PFS was 8.8 months in the 90 mg dose group, and 11.1 months in the 180mg dose group. The most common adverse reactions, were nausea, diarrhea, fatigue, cough, and headache. The most common serious adverse reactions were pneumonia and pneumonitis.
The authors concluded that treatment with ALUNBRIG® resulted in significant response rates and improved Progression Free Survival, with acceptable toxicity profile. The recommended dosing regimen of ALUNBRIG® is 90 mg orally once daily for the first 7 days and if tolerated, the dose is increased to 180 mg orally once daily.
Kim D-W, Tiseo M, Ahn M-J, et al. Brigatinib (BRG) in patients (pts) with crizotinib (CRZ)-refractory ALK+ non-small cell lung cancer (NSCLC): First report of efficacy and safety from a pivotal randomized phase (ph) 2 trial (ALTA). J Clin Oncol. 2016;34 (suppl; abstr 9007).



