SUMMARY: Prostate cancer is the most common cancer in American men with the exclusion of skin cancer and 1 in 7 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 161,360 new cases of Prostate cancer will be diagnosed in 2017 and 26,730 men will die of the disease. The development and progression of prostate cancer is driven by androgens. Androgen Deprivation Therapy (ADT) has therefore been the cornerstone of treatment of advanced prostate cancer and is the first treatment intervention for hormone sensitive prostate cancer. For patients with Castrate Resistant Prostate Carcinoma (CRPC), several agents have been proven to improve Overall Survival and they include, TAXOTERE® (Docetaxel), JEVTANA® (Cabazitaxel), ZYTIGA® (Abiraterone acetate), XTANDI® (Enzalutamide), XOFIGO® (Radium-223), and PROVENGE® (Sipuleucel-T).
The Systemic Therapy in Advancing or Metastatic Prostate Cancer: Evaluation of Drug Efficacy (STAMPEDE) trial is an ongoing study and uses a novel multiarm, multistage (MAMS) platform design, to test whether the addition of further treatments to ADT improves Overall Survival, when used in first-line setting, for patients with hormone sensitive, locally advanced or metastatic prostate cancer. This group previously reported that there was a significantly improved Overall Survival with the addition of TAXOTERE® to initial ADT. This is presently the standard of care for appropriate patients with prostate cancer, who had not received prior hormone therapy. The barriers to chemo-hormonal therapy with TAXOTERE® include advanced patient age, poor Performance Status, comorbidities, patient preferences, as well as potential life threatening toxicities associated with TAXOTERE®.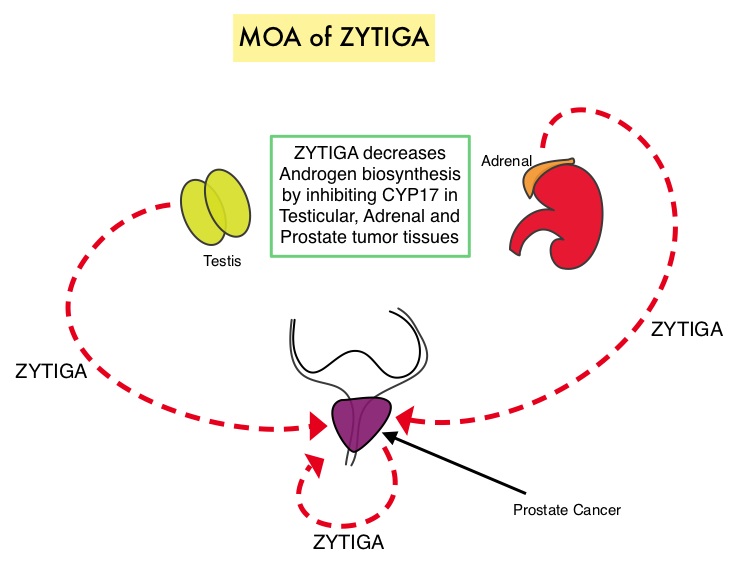
ZYTIGA® is a selective, irreversible inhibitor of CYP 17A1 enzyme and decreases androgen biosynthesis in the testes, adrenal glands, and prostate-tumor tissue. Combining a CYP17A1 inhibitor such as ZYTIGA® with Androgen Deprivation Therapy is a more effective way of androgen depletion than with Orchiectomy or GnRH analogues alone. In this analysis, the STAMPEDE trial evaluated Overall Survival outcomes, with the earlier use of ZYTIGA®, in men with high risk, hormone sensitive prostate cancer, who were initiating long-term Androgen Deprivation Therapy.
A total of 1917 patients were randomly assigned patients in a 1:1 ratio to receive Androgen Deprivation Therapy (ADT) alone (N=957) or ADT plus ZYTIGA® (N=960), administered at 1000 mg PO daily and prednisolone 5 mg PO daily (combination therapy). ADT was given for at least 2 years. Eligible patients had prostate cancer that was newly diagnosed and metastatic, node-positive, or high-risk locally advanced disease or prostate cancer that was previously treated with radical surgery or radiotherapy and was now relapsing with high-risk features. Patients with locally advanced disease could also receive radiation therapy in addition to ADT. Radiotherapy was mandated for patients with N0M0 disease and encouraged for those with stage N+M0 disease. The median age was 67 years, and the median PSA level was 53 ng/ml. Approximately 52% of the patients had metastatic disease, 20% had node-positive or node-indeterminate, non-metastatic disease, and 28% had node-negative, non-metastatic disease. Majority of the patients (95%) had newly diagnosed disease. The median follow up was 40 months.
There was a significant survival advantage with combination therapy with a 3-year Overall Survival of 83% with ADT plus ZYTIGA® compared with 76% with ADT alone group (HR=0.63; P<0.001). This meant a 37% reduction in the risk of death with the ZYTIGA® combination treatment. Further, the combination treatment reduced the risk of relapse by 71% (HR=0.29; P<0.001), and also reduced the risk of symptomatic skeletal events by 54% (HR=0.46; P<0.001), compared with ADT alone. Treatment overall was well tolerated.
It was concluded that ADT plus ZYTIGA® and prednisolone results in significantly higher rates of Overall Survival as well as Failure-Free Survival, compared with ADT alone, among men with hormone sensitive, locally advanced or metastatic prostate cancer. Interestingly, the LATITUDE trial showed similar findings in newly diagnosed, metastatic, hormone sensitive prostate cancer patients (June 4, 2017DOI: 10.1056/NEJMoa1704174). The results of both STAMPEDE and LATITUDE trials will very likely change practice patterns and will become the new standard of care for this patient group. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. James ND, de Bono JS, Spears MR, et al. for the STAMPEDE Investigators. June 3, 2017DOI: 10.1056/NEJMoa1702900