SUMMARY: Myelofibrosis is a MyeloProliferative Neoplasm (MPN) characterized by a ineffective hematopoiesis, progressive fibrosis of the bone marrow and potential for leukemic transformation. This stem cell disorder is Philadelphia Chromosome negative and manifestations include anemia, splenomegaly and related symptoms such as abdominal distension and discomfort with early satiety. Cytokine driven debilitating symptoms such as fatigue, fever, night sweats, weight loss, pruritus and bone or muscle pain can further impact an individual’s quality of life. Myelofibrosis can be primary (PMF) or secondary to Polycythemia Vera (PV) or Essential Thrombocythemia (ET). 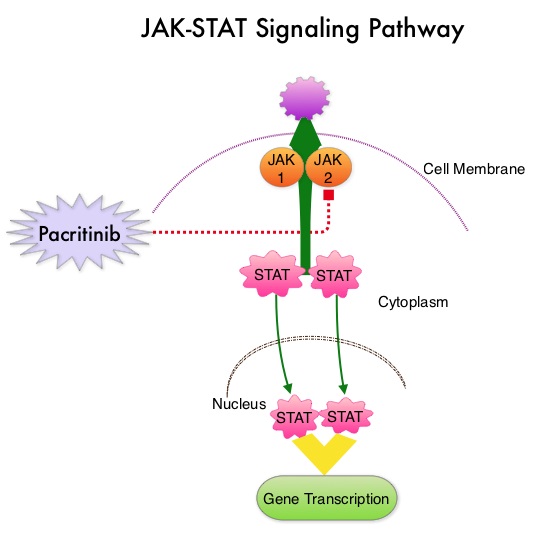 The JAK-STAT signaling pathway has been implicated in the pathogenesis of Myelofibrosis. This pathway normally is responsible for passing information from outside the cell through the cell membrane to the DNA in the nucleus, for gene transcription. Janus Kinase (JAK) family of tyrosine kinases are cytoplasmic proteins and include JAK1, JAK2, JAK3 and TYK2. JAK1 helps propagate the signaling of inflammatory cytokines whereas JAK2 is essential for growth and differentiation of hematopoietic stem cells. These tyrosine kinases mediate cell signaling by recruiting STAT’s (Signal Transducer and Activator of Transcription), with resulting modulation of gene expression. In patients with MPN, the aberrant myeloproliferation is the result of dysregulated JAK2-STAT signaling as well as excess production of inflammatory cytokines associated with this abnormal signaling. These cytokines contribute to the symptoms often reported by patients with MF. JAK2 mutations such as JAK2 V617F are seen in approximately 60% of the patients with PMF and ET and 95% of patients with PV. Unlike CML where the BCR-ABL fusion gene triggers the disease, JAK2 mutations are not initiators of the disease and are not specific for MPN. Further, several other genetic events may contribute to the abnormal JAK2-STAT signaling.
The JAK-STAT signaling pathway has been implicated in the pathogenesis of Myelofibrosis. This pathway normally is responsible for passing information from outside the cell through the cell membrane to the DNA in the nucleus, for gene transcription. Janus Kinase (JAK) family of tyrosine kinases are cytoplasmic proteins and include JAK1, JAK2, JAK3 and TYK2. JAK1 helps propagate the signaling of inflammatory cytokines whereas JAK2 is essential for growth and differentiation of hematopoietic stem cells. These tyrosine kinases mediate cell signaling by recruiting STAT’s (Signal Transducer and Activator of Transcription), with resulting modulation of gene expression. In patients with MPN, the aberrant myeloproliferation is the result of dysregulated JAK2-STAT signaling as well as excess production of inflammatory cytokines associated with this abnormal signaling. These cytokines contribute to the symptoms often reported by patients with MF. JAK2 mutations such as JAK2 V617F are seen in approximately 60% of the patients with PMF and ET and 95% of patients with PV. Unlike CML where the BCR-ABL fusion gene triggers the disease, JAK2 mutations are not initiators of the disease and are not specific for MPN. Further, several other genetic events may contribute to the abnormal JAK2-STAT signaling.
Pacritinib is a potent JAK2 inhibitor, without significant JAK1 inhibition. Preliminary studies have shown minimal myelosuppression with Pacritinib. JAKAFI® (Ruxolitinib) is a potent JAK1 and JAK2 inhibitor and is not safe for patients with low platelet counts. PERSIST-1 is a phase III study in which 327 patients with intermediate or high-risk Primary MyeloFibrosis (PMF), Post–Polycythemia Vera MF or Post–Essential Thrombocythemia MF were enrolled. Patients were randomized in a 2:1 ratio to receive Pacritinib 400 mg once daily (N=220) or Best Available Therapy (BAT) excluding JAKAFI® (N=107). Patients in the BAT group received Erythropoietin Stimulating Agents, Immunomodulatory drugs such as THALOMID® (Thalidomide), REVLIMID® (Lenalidomide) and Hydroxyurea. Because patients with very low platelet counts were enrolled in this study, JAKAFI® therapy was excluded, as JAKAFI® is not considered safe for patients with low platelet count. Approximately 32% of the patients had a platelet count of less than 100,000/µL and 15% had a platelet count of less than 50,000/ µL. About 75% of the patients were JAK2V617F positive. The median duration of treatment was 16.2 months in the Pacritinib group and 5.9 months in the BAT group. The primary endpoint was the proportion of patients achieving 35% or more reduction in the spleen volume at 24 weeks. Secondary endpoints included the proportion achieving 50% or more reduction in MyeloProliferative Neoplasm symptom score at 24 weeks.
At 24 weeks of treatment, 19.1% of patients in the Pacritinib group experienced 35% or more reduction in spleen volume compared to 4.7% in the Best Available Therapy (BAT) group (P=0.0003). This benefit was even more so in the subgroup of patients with the lowest platelet counts (less than 50,000/ µL), with 33.3% in the Pacritinib group demonstrating spleen volume reduction, compared to none in the BAT group. Patients in the Pacritinib arm were much more likely to experience more than a 50% reduction in symptoms, compared to BAT group at 24 weeks (24.5% vs 6.5%; P<0.0001), with significant improvements in fatigue, early satiety, abdominal discomfort, pruritus, night sweats and bone pain. This symptom improvement was noted by 4-8 weeks. Approximately 25% of the patients in the Pacritinib group achieved transfusion independence compared with none in the control group (P=0.043). The most common adverse events associated with Pacritinib were diarrhea, nausea and vomiting.
The authors concluded that Pacritinib significantly reduces spleen volume and Myelofibrosis associated symptoms, and fulfills an unmet need for Myelofibrosis patients with low platelet count, in addition to achieving RBC transfusion independence. Results of the PERSIST-1 phase III study of pacritinib (PAC) versus best available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF), or post-essential thrombocythemia-myelofibrosis (PET-MF). Mesa RA, Egyed M, Szoke A, et al. J Clin Oncol 33, 2015 (suppl; abstr LBA7006)
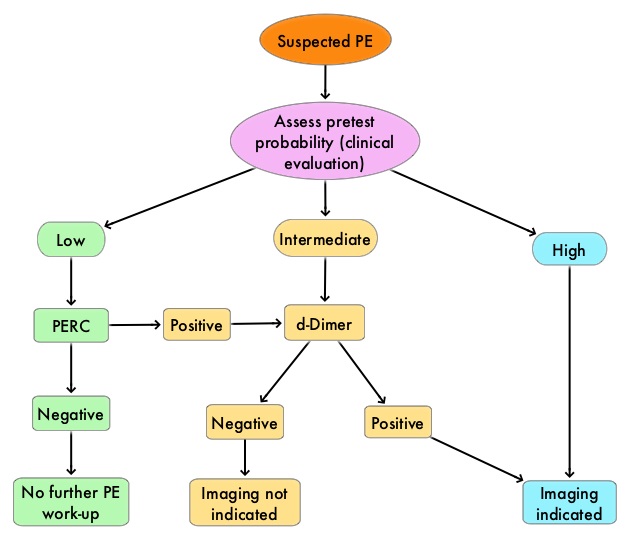
 There has also been an overuse of CT imaging and plasma d-Dimer measurement, without improvement in care, but rather harming the patient and increasing expenditure. The validated clinical decision tools, in addition to physician’s clinical judgment include the Wells and Geneva Scoring System. The PERC (see table) criteria includes 8 elements, which if absent in low risk patients rules out PE. These practice guidelines were developed to provide practical advice, based on the best available evidence.
There has also been an overuse of CT imaging and plasma d-Dimer measurement, without improvement in care, but rather harming the patient and increasing expenditure. The validated clinical decision tools, in addition to physician’s clinical judgment include the Wells and Geneva Scoring System. The PERC (see table) criteria includes 8 elements, which if absent in low risk patients rules out PE. These practice guidelines were developed to provide practical advice, based on the best available evidence.