SUMMARY: The FDA on September 22, 2017, granted accelerated approval to OPDIVO® (Nivolumab) for the treatment of HepatoCellular Carcinoma (HCC), in patients who have been previously treated with NEXAVAR® (Sorafenib). The American Cancer Society estimates that about 41,000 new cases of primary liver cancer will be diagnosed in the US for 2017 and 29,000 patients will die of their disease. Liver cancer is seen more often in men than in women and the incidence has more than tripled since1980. This increase has been attributed to the higher rate of hepatitis C virus (HCV) infection among baby boomers (born between 1945 through 1965). Obesity and type II diabetes have also likely contributed to the trend. Other risk factors include alcohol, which increases liver cancer risk by about 10% per drink per day, and tobacco use, which increases liver cancer risk by approximately 50%. HCC is the second most common cause of cancer-related deaths worldwide. NEXAVAR® (Sorafenib) was approved by the FDA in 2007 for the treatment of unresectable, HepatoCellular Carcinoma (HCC). Patients with advanced HCC who progress on NEXAVAR®, have a poor prognosis with limited treatment options.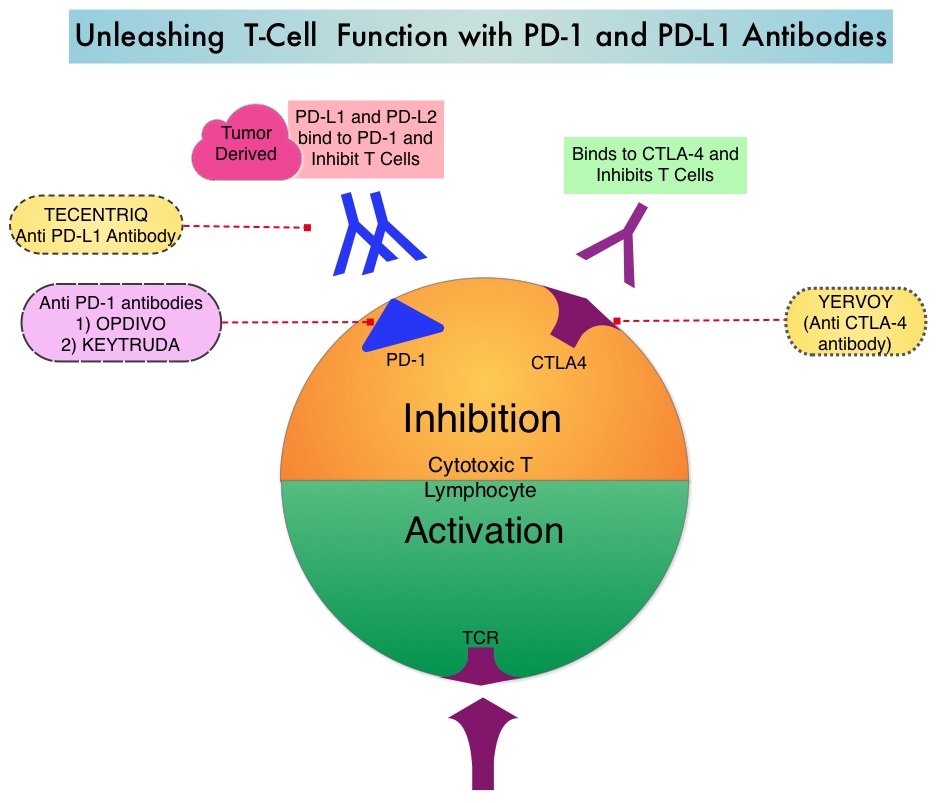
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Under normal circumstances, Immune checkpoints or gate keepers inhibit intense immune responses by switching off the T cells of the immune system and can thereby suppress antitumor immunity. These Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. Immune check point inhibitors are antibodies that target the immune checkpoint receptors and unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. OPDIVO® (Nivolumab) is an immune checkpoint PD-1 (Programmed cell Death 1) targeted, fully human, immunoglobulin G4 monoclonal antibody that demonstrated significant activity in HepatoCellular Carcinoma (HCC).
The approval of OPDIVO® in HCC was based on a 154-patient subgroup of CHECKMATE-040 study, which is a multicenter, open-label trial, conducted in patients with HCC and Child-Pugh A cirrhosis, who had progressed on or were intolerant to NEXAVAR®. Enrolled patients included patients without active viral hepatitis, patients with either active HBV-Hepatitis B Virus (31%) or HCV- Hepatitis C Virus (21%). Patients with active co-infection with HBV and HCV or with Hepatitis D Virus infection were excluded. Patients received OPDIVO® 3 mg/kg by IV infusion every 2 weeks. The median age was 64 years and 80% of patients were male. The primary endpoints were safety/tolerability and Objective Response Rate (ORR) by investigator and Blinded Independent Central Review. Secondary endpoints included Duration of Response, Disease Control Rate and Overall Survival.
The confirmed Objective Response Rate as assessed by Independent Central Review was 19 % with 3 Complete Responses and 19 partial responses(13%) and 41% of the patients had stable disease. Response duration ranged from 3 months to over 38 months and 91% of responders had responses lasting 6 months or longer and 55% of the patients had responses lasting 12 months or longer. Overall responses were noted regardless of tumor cell PD-L1 expression and chronic hepatitis virus infection. Common adverse reactions included fever, rash, musculoskeletal pain, nausea, diarrhea, cough and dyspnea. There was a higher incidence of elevations in transaminases and bilirubin levels noted.
It was concluded that OPDIVO® demonstrated durable responses with long-term survival and favorable safety in patients with advanced HCC. This FDA approval provides a new treatment option for appropriate patients with HCC, who progress on prior systemic therapy. STIVARGA® (Regorafenib) is another agent presently available for this subset of patients. Nivolumab (nivo) in sorafenib (sor)-naive and -experienced pts with advanced hepatocellular carcinoma (HCC): CheckMate 040 study. Crocenzi TS, El-Khoueiry AB, Yau TC, et al. J Clin Oncol. 2017;35 (suppl; abstr 4013).