SUMMARY: Prostate cancer is the most common cancer in American men with the exclusion of skin cancer, and 1 in 9 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 164,690 new cases of prostate cancer will be diagnosed in 2018 and 29,430 men will die of the disease. Prostate cancer is driven by Androgen Receptor (AR) and its signaling pathways. Initial treatment strategies for patients with metastatic prostate cancer include lowering the levels of circulating androgens with medical or surgical castration or blocking the binding of androgens to the androgen receptor. Upon progression, {described as Castrate Resistant Prostate Cancer (CRPC), as these tumors are not androgen independent and continue to rely on Androgen Receptor signaling}, Androgen Receptor Signaling (ARS) inhibitors, such as ZYTIGA ® (Abiraterone acetate) and XTANDI® (Enzalutamide), and Taxanes such as TAXOTERE® (Docetaxel) and JEVTANA® (Cabazitaxel) are the most widely used drug classes in the United States. ZYTIGA® inhibits CYP17A1 enzyme and depletes adrenal and intratumoral androgens, thereby impairing AR signaling. XTANDI® competes with Testosterone and Dihydrotestosterone and avidly binds to the Androgen Receptor, thereby inhibiting AR signaling, and in addition inhibits translocation of the AR into the nucleus and thus inhibits the transcriptional activities of the AR. About 20-40% of the patients do not respond to these newer agents (ARS inhibitors), and even those who respond will invariably develop resistance to these drugs.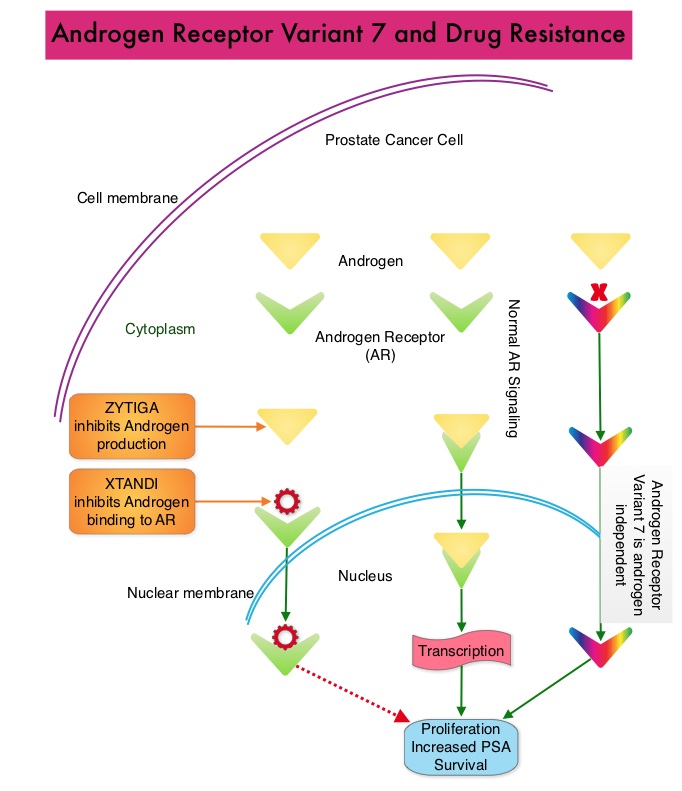
ARS inhibitors are often the preferred choice for the first-line treatment of metastatic CRPC (mCRPC). In clinical practice, the majority of patients with mCRPC who progress on first-line ARS inhibitor receive a second ARS inhibitor, as there are no formal guidelines on how best to sequence these agents after progression on first-line ARS inhibition. Resistance to ARS inhibitors has been attributed to persistent AR signaling by variant forms of Androgen Receptor, generated through somatic mutation or aberrant RNA splicing. Androgen Receptor splice Variant 7 (AR-V7) is the most widely studied and can be detected in the CTCs (Circulating Tumor Cells). AR-V7 does not have the domain to bind androgens and may be associated with resistance to XTANDI®. Further AR-V7 is constitutively active and can independently activate transcription factors and therefore is not effected by androgen depleting agents including ZYTIGA®.
A critical unmet need is a test that can provide guidance on selecting appropriate therapy in the second and later lines, for this patient group. Accurately detecting a splice variant of the Androgen Receptor protein (AR-V7) in the nucleus of Circulating Tumor Cells (CTCs) using peripheral blood (liquid biopsy), would enable treating physicians to confidently decide whether patients treated with an ARS inhibitor, upon progression, would benefit from a second ARS inhibitor or should be switched to chemotherapy. This study focused on the patients receiving second-line treatment, to determine if an assay for the nuclear-localized AR-V7 protein in CTCs can be used to determine treatment for mCRPC.
The authors conducted this independent, multicenter cohort study to determine whether a validated assay for the nuclear-localized Androgen Receptor splice Variant 7 (AR-V7) protein in Circulating Tumor Cells (CTCs) can be used as a treatment-selection marker for metastatic CRPC, and whether it can determine Overall Survival difference among patients with mCRPC treated with Taxanes versus ARS inhibitors. This blinded correlative study included 142 patients with histologically confirmed mCRPC. Blood samples were obtained prior to administration of ARS inhibitors (N=70) or Taxanes (N=72), as a second-line therapy or later, for progressing mCRPC . Mean age was 69.5 years. Seventy (N=70) patients were designated as high risk by conventional prognostic factors. The Primary outcome measure was Overall Survival after treatment with an ARS inhibitor or Taxane, in relation to pretreatment AR-V7 status.
It was noted that the presence of AR-V7 protein in CTCs was associated with a greater benefit from Taxane treatment whereas outcomes in AR-V7 negative patients were better with ARS inhibitor treatment. The median survival of patients with AR-V7 negative CTCs was 19.8 months for those treated with an ARS inhibitor and 12.8 months for those treated with a Taxane (P=0.05). In contrast, among patients with AR-V7 positive CTCs, those receiving Taxanes had longer observed median survival times compared to those treated with ARS inhibitors (14.3 vs 7.3 months; HR=0.62; P=0.25). The observed difference was not statistically significant and may have been attributable to the small sample size.
The authors concluded that this study suggests that nuclear-localized AR-V7 protein in Circulating Tumor Cells (liquid biopsy test) can identify patients who may live longer with Taxane chemotherapy versus ARS inhibitor treatment. This tailored treatment approach addresses a critical unmet need by predicting and enabling selection of therapy that can improve median survival. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. Scher HI, Graf RP, Schreiber NA, et al. JAMA Oncol. Published online June 28, 2018. doi:10.1001/jamaoncol.2018.1621