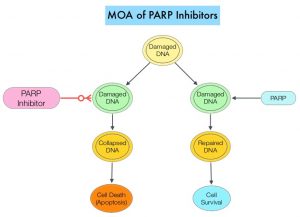
SUMMARY: Radiation Therapy involves the use of X-Rays, Gamma rays and charged particles for cancer treatment. External Beam Radiation Therapy (EBRT) is most often delivered using a linear accelerator in the form of Photon beams (either X-rays or Gamma rays). Photons have no mass and are packets of energy of an electromagnetic wave. Electrons and Protons are charged particles and Electrons are considered light particles whereas Protons are considered heavy particles. Electron beams are used to irradiate skin and superficial tumors, as they are unable to penetrate deep into the tissues. The different types of External Beam Radiation Treatments include 3-Dimensional Conformal Radiation Therapy (3D-CRT) meant to deliver radiation to very precisely shaped target areas, IMRT or Intensity Modulated Radiation Therapy which allows different areas of a tumor or nearby tissues to receive different doses of radiation, Image Guided Radiation Therapy (IGRT) which allows reduction in the planned volume of tissue to be treated, as changes in a tumor size are noted during treatment, Stereotactic RadioSurgery (SRS) which can deliver one or more high doses of radiation to a small tumor and Stereotactic Body Radiation Therapy (SBRT) or CYBERKNIFE® which is similar to SRS but also takes the normal motion of the body into account while treating malignancies involving the lung and liver.
Proton beams unlike Photons, enter the skin and travel through the tissues and deposit much of their energy at the end of their path (known as the Bragg peak), and deposit less energy along the way. This is unlike Photons which deposit energy all along the path through the tissues and the deposited dose decreases with increasing depth. As a result, with Proton beam therapy, normal tissues are exposed to less radiation compared with Photons. Despite this advantage, tissue heterogeneity such as organ motion, tumor volume changes during treatment can have a significant negative impact on target coverage for Proton beam therapy and can result in damage to the surrounding tissues and potential complications. It is well established that there is significant benefit for Proton beam therapy in certain pediatric malignancies.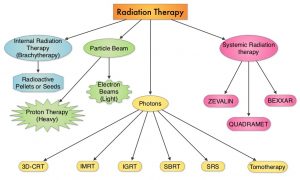
Curative treatment with concurrent chemoradiotherapy is the standard of care for many nonmetastatic, locally advanced cancers. This treatment modality however is associated with substantial morbidity. Proton therapy as component of concurrent chemoradiotherapy might be able to reduce treatment related toxicity and achieve comparable cancer control outcomes, compared with conventional Photon radiotherapy, by reducing the radiation dose to normal tissues. There are however limited data comparing results of Proton chemoradiotherapy with Photon chemoradiotherapy, and Proton therapy remains unproven in this treatment setting. The objective of this study was to assess whether Proton therapy in the setting of concurrent chemoradiotherapy is associated with fewer hospitalizations or other adverse events and similar Disease-free and Overall Survival, compared with concurrent Photon chemoradiotherapy.
In this large single-institution, nonrandomized, comparative effectiveness, retrospective analysis, 1483 adult patients with nonmetastatic, locally advanced cancer, treated with concurrent chemoradiotherapy with curative intent were included. Three hundred ninety-one patients (N=391) received Proton therapy and 1092 patients received Photon therapy. Common tumor sites included head and neck, lung, brain, esophagus/gastric, rectum, and pancreas. The median patient age was 62 years, but patients treated with Protons were significantly older with a median age of 66 years versus 61 years, had less favorable Charlson-Deyo comorbidity scores and had lower integral radiation dose to tissues outside the target. Ninety three percent (93%) of patients in the Photon therapy group were treated with Intensity-Modulated Radiotherapy (IMRT). Baseline ECOG Performance Status was similar between the two treatment cohorts. The Primary end point was 90-day adverse events associated with unplanned hospitalizations (CTCAE version 4 – Grade 3 or more). Secondary end points included ECOG performance status decline during treatment, 90-day adverse events of at least Grade 2 that limit instrumental activities of daily living, and Disease-Free and Overall Survival. The data on adverse events and survival were gathered prospectively.
It was noted that Proton chemoradiotherapy was associated with a significantly lower relative risk of 90-day adverse events of at least Grade 3 (P=0.002), significantly lower relative risk of 90-day adverse events of at least Grade 2 (P=0.006), and decline in Performance Status during treatment (P<0.001). Proton chemoradiotherapy was associated with a two-thirds reduction in adverse events associated with unplanned hospitalizations. At a median follow up of 3.7 years for the Proton cohort and 4.2 years for the Photon cohort, there was no difference in Disease-Free or Overall Survival.
It was concluded from this analysis that in adults with locally advanced cancer, Proton chemoradiotherapy was associated with significantly reduced acute adverse events that caused unplanned hospitalizations, with similar Disease-Free and Overall Survival, compared to Photon therapy.
Comparative Effectiveness of Proton vs Photon Therapy as Part of Concurrent Chemoradiotherapy for Locally Advanced Cancer. Baumann BC, Mitra N, Harton JG, et al. Jama Oncol. 2020;6:237-246.