SUMMARY: The FDA on May 15, 2020, granted accelerated approval to RUBRACA® (Rucaparib) for patients with deleterious BRCA mutation (germline and/or somatic)-associated metastatic Castration-Resistant Prostate Cancer (mCRPC), who have been treated with Androgen Receptor-directed therapy and a taxane-based chemotherapy. Prostate cancer is the most common cancer in American men with the exclusion of skin cancer, and 1 in 9 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 191,930 new cases of prostate cancer will be diagnosed in 2020 and 33,330 men will die of the disease.
The development and progression of Prostate cancer is driven by androgens. Androgen Deprivation Therapy (ADT) or testosterone suppression has therefore been the cornerstone of treatment of advanced Prostate cancer and is the first treatment intervention. Androgen Deprivation Therapies have included bilateral orchiectomy or Gonadotropin Releasing Hormone (GnRH) analogues, with or without first generation Androgen Receptor (AR) inhibitors such as CASODEX® (Bicalutamide), NILANDRON® (Nilutamide) and EULEXIN® (Flutamide) or with second-generation, anti-androgen agents, which include, ZYTIGA® (Abiraterone), XTANDI® (Enzalutamide) and ERLEADA® (Apalutamide). ZYTIGA® inhibits CYP17A1 enzyme and depletes adrenal and intratumoral androgens, thereby impairing AR signaling. XTANDI® and ERLEADA® compete with Testosterone and Dihydrotestosterone and avidly bind to the Androgen Receptor, thereby inhibiting AR signaling, and in addition inhibit translocation of the AR into the nucleus and thus inhibits the transcriptional activities of the AR. Approximately 10-20% of patients with advanced Prostate cancer will progress to Castration Resistant Prostate Cancer (CRPC) within five years during ADT, and over 80% of these patients will have metastatic disease at the time of CRPC diagnosis. The estimated mean survival of patients with CRPC is 9-36 months, and there is therefore an unmet need for new effective therapies.
DNA damage is a common occurrence in daily life by UV light, ionizing radiation, replication errors, chemical agents, etc. This can result in single and double strand breaks in the DNA structure which must be repaired for cell survival. The two vital pathways for DNA repair in a normal cell are BRCA1/BRCA2 and PARP. The PARP (Poly ADP Ribose Polymerase), family of enzymes include, PARP1, PARP2 and PARP3. BRCA1 and BRCA2 genes recognize and repair double strand DNA breaks via Homologous Recombination (HR) pathway. Homologous Recombination is a type of genetic recombination, and is a DNA repair pathway utilized by cells to accurately repair DNA double-stranded breaks during the S and G2 phases of the cell cycle, and thereby maintain genomic integrity. Homologous Recombination Deficiency (HRD) is noted following mutation of genes involved in HR repair pathway. At least 15 genes are involved in the Homologous Recombination Repair (HRR) pathway including BRCA1, BRCA2 and ATM genes. The BRCA1 gene is located on the long (q) arm of chromosome 17 whereas BRCA2 is located on the long arm of chromosome 13. BRCA1 and BRCA2 are tumor suppressor genes and functional BRCA proteins therefore repair damaged DNA, and play an important role in maintaining cellular genetic integrity. They regulate cell growth and prevent abnormal cell division and development of malignancy.
Recently published data has shown that deleterious germline and/or somatic mutations in BRCA1, BRCA2, ATM, or other Homologous Recombination DNA-repair genes, are present in about 25% of patients with advanced prostate cancer, including mCRPC. Mutations in BRCA1 and BRCA2 also account for about 20-25% of hereditary breast cancers, about 5-10% of all breast cancers, and 15% of ovarian cancers. BRCA mutations can either be inherited (Germline) and present in all individual cells or can be acquired and occur exclusively in the tumor cells (Somatic). Somatic mutations account for a significant portion of overall BRCA1 and BRCA2 aberrations. Loss of BRCA function due to frequent somatic aberrations likely deregulates HR pathway, and other pathways then come in to play, which are less precise and error prone, resulting in the accumulation of additional mutations and chromosomal instability in the cell, with subsequent malignant transformation. HRD therefore indicates an important loss of DNA repair function. PARP is a related enzymatic pathway that repairs single strand breaks in DNA. In a BRCA mutant, the cancer cell relies solely on PARP pathway for DNA repair. In the presence of a PARP inhibitor, there is synthetic lethality because of the loss of both repair pathway genes, leading to cell death. Thus PARP inhibitors are only harmful to cancer cells.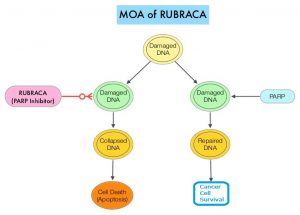
RUBRACA® is an oral, small molecule inhibitor of Poly-Adenosine diphosphate [ADP] Ribose Polymerase (PARP), presently approved by the FDA for ovarian, fallopian tube or primary peritoneal cancers. This recent FDA approval for prostate cancer patients was based on TRITON2, which is an ongoing international, multicenter, open-label, single arm, Phase II trial, in patients with BRCA-mutated mCRPC, who had been treated with Androgen Receptor-directed therapy and taxane-based chemotherapy. In this study, 115 mCRPC patients with either germline or somatic BRCA mutations were enrolled, of whom 62 patients had measurable disease at baseline. Patients received RUBRACA® 600 mg orally twice daily and concomitant GnRH analog or had prior bilateral orchiectomy. Treatment was continued until disease progression or unacceptable toxicity. The median age was 73 years, majority of patients had an ECOG performance status of 0 or 1, 18% of patients had lung metastases, 21% had liver metastases, 24% had metastases to lymph nodes alone and 40% had 10 or more bone lesions at baseline. The major efficacy outcomes of the study were Objective Response Rate (ORR) and Duration of Response (DOR) in the 62 patients with measurable disease. The median duration of follow up was 13.1 months
The confirmed ORR was 44% and the median DOR was not evaluable. Fifty six percent (56%) of patients with confirmed Objective Responses had a DOR of 6 months or more.
It was concluded that RUBRACA® demonstrates promising efficacy in patients with mCRPC with deleterious BRCA mutations. TRITON3 study is evaluating RUBRACA® versus physician’s choice of second-line AR-directed therapy or Docetaxel, in chemotherapy-naïve patients with mCRPC and alterations in BRCA1/2, who progressed on one prior AR-directed therapy.
ESMO 2019: Preliminary Results from the TRITON2 Study of Rucaparib in Patients with DNA Damage Repair-deficient mCRPC: Updated Analyses. Abida W, Campbell D, Patnaik A, et al. 2019 ESMO Annual Meeting, #ESMO19, 27 Sept – 1 Oct 2019 in Barcelona, Spain.