SUMMARY: The FDA on May 29, 2020 approved CYRAMZA® (Ramucirumab) in combination with TARCEVA® (Erlotinib) for first-line treatment of metastatic Non-Small Cell Lung Cancer (NSCLC) with Epidermal Growth Factor Receptor (EGFR) Exon 19 deletions or Exon 21 (L858R) mutations. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR mutations and 90% of these mutations are either Exon 19 deletions or L858R substitution mutation in Exon 21. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA®, IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to the most common, acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients. Previously published data from the Phase III FLAURA study showed that first-line treatment with third generation TKI, TAGRISSO® (Osimertinib), was superior to first-line treatment with other first and second generation TKI’s, in patients with EGFR-mutated NSCLC. However, widespread use of TAGRISSO® has led to acquired resistance. Novel treatment approaches combining TKI’s with other targeted therapies are therefore needed.
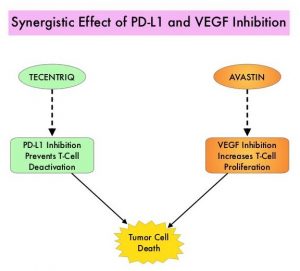
CYRAMZA® is a recombinant human monoclonal IgG1 antibody that binds to the human Vascular Endothelial Growth Factor Receptor- 2 (VEGFR-2), preventing the interaction of VEGFR-2 with its ligands. TARCEVA® is a first generation EGFR TKI. Preclinical and clinical data strongly support dual blockade of the EGFR and VEGF pathways in EGFR-mutated metastatic NSCLC.
RELAY is an International, double-blind, Phase III trial, which included 449 eligible patients who had Stage IV NSCLC, with an EGFR Exon 19 deletion (ex19del) or Exon 21 substitution (L858R) mutation, and with no CNS metastases. Enrolled patients were randomly assigned in a 1:1 ratio to receive TARCEVA® 150 mg orally daily plus CYRAMZA® 10 mg/kg IV once every 2 weeks (N=224) or TARCEVA® plus a matching placebo (N=225). Patients were stratified by sex, EGFR mutation type, and EGFR testing methodology. The Primary endpoint was Progression Free Survival (PFS) and key Secondary endpoints included Safety, Overall Response Rate (ORR), Duration of Response, and Overall Survival (OS).
At a median follow up of 20.7 months, PFS was significantly longer in the TARCEVA® plus CYRAMZA® group compared to TARCEVA® plus placebo group (19.4 months versus 12.4 months respectively; HR=0.59; P<0.0001). This benefit was observed regardless of tumor type, and was consistent across Exon 19 and Exon 21 subgroups. The ORR was similar between the CYRAMZA® and placebo groups (76% versus 75%), but the median Duration of Response was longer in the CYRAMZA® group, compared with the placebo group (18 months versus 11 months). The OS data were not mature at the time of final PFS analysis and the median time to the second disease progression (PFS2) was not yet reached. However, interim results indicated that PFS2 was longer in the CYRAMZA® group compared to the placebo group (HR = 0.69) suggesting that PFS benefits with CYRAMZA® were preserved beyond first progression, indicating that possibility of OS benefit. Upon progression, T790M resistance mutations were detected in 43% of patients who received CYRAMZA®, and in 47% of patients who received placebo. The most common adverse events in the TARCEVA® plus CYRAMZA® combination included infections, stomatitis, hypertension, proteinuria, alopecia, epistaxis and peripheral edema.
It was concluded that TARCEVA® plus CYRAMZA® demonstrated superior PFS compared with TARCEVA® plus placebo, in treatment naïve patients with EGFR-mutated metastatic NSCLC. The combination of TARCEVA® plus CYRAMZA® will be a new additional treatment option for this patient group.
Ramucirumab plus Erlotinib in Patients with Untreated, EGFR-mutated, Advanced Non-Small-Cell Lung Cancer (RELAY): A Randomised, Double-blind, Placebo-Controlled, Phase 3 trial. Nakagawa K, Garon EB, Seto T, et al. Lancet Oncol. 2019;20:1655-1669.