
The FDA on April 21, 2020 expanded the indication of IMBRUVICA® to include its combination with RITUXAN® (Rituximab) for the initial treatment of adult patients with Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL). IMBRUVICA® is a product of Pharmacyclics LLC.
Author: RR
PEMAZYRE® (Pemigatinib)
The FDA on April 20, 2020, granted accelerated approval to PEMAZYRE® for the treatment of adults with previously treated, unresectable locally advanced or metastatic Cholangiocarcinoma with a Fibroblast Growth Factor Receptor 2 (FGFR2) fusion or other rearrangement, as detected by an FDA-approved test. PEMAZYRE® is a product of Incyte Corporation.
FDA Approves TUKYSA® for HER2+ Breast Cancer
SUMMARY: The FDA on April 17, 2020, approved TUKYSA® (Tucatinib) in combination with Trastuzumab and XELODA® (Capecitabine), for adult patients with advanced unresectable or metastatic HER2-positive breast cancer, including patients with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting. Breast cancer is the most common cancer among women in the US and about 1 in 8 women (13%) will develop invasive breast cancer during their lifetime. Approximately 276,480 new cases of invasive female breast cancer will be diagnosed in 2020 and about 42,170 women will die of the disease.
The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. Patients with HER2-positive metastatic breast cancer are often treated with anti-HER2 targeted therapy along with chemotherapy, irrespective of hormone receptor status, and this has resulted in significantly improved treatment outcomes. HER2-targeted therapies include HERCEPTIN® (Trastuzumab), TYKERB® (Lapatinib), PERJETA® (Pertuzumab) and KADCYLA® (ado-Trastuzumab emtansine). Dual HER2 blockade with HERCEPTIN® and PERJETA®, given along with chemotherapy (with or without endocrine therapy), as first line treatment, in HER2 positive metastatic breast cancer patients, was shown to significantly improve Progression Free Survival (PFS) as well as Overall Survival (OS). The superior benefit with dual HER2 blockade has been attributed to differing mechanisms of action and synergistic interaction between HER2 targeted therapies. Patients progressing on Dual HER2 blockade often receive KADCYLA® which results in an Objective Response Rate (ORR) of 44% and a median PFS of 9.6 months, when administered after HERCEPTIN® and a taxane. There is however no standard treatment option for this patient population following progression on KADCYLA®.
It is estimated that close to 50% of patients with HER2-positive metastatic breast cancer develop brain metastases. Systemic HER2-targeted agents, including Tyrosine Kinase Inhibitors, as well as chemotherapy have limited antitumor activity in the brain. Local therapeutic interventions for brain metastases include neurosurgical resection and Stereotactic or Whole-Brain Radiation Therapy.
TUKYSA® (Tucatinib) is an oral Tyrosine Kinase Inhibitor that is highly selective for the kinase domain of HER2 with minimal inhibition of Epidermal Growth Factor Receptor. In a Phase 1b dose-escalation trial, TUKYSA® in combination with HERCEPTIN® and XELODA® (Capecitabine) showed encouraging antitumor activity in patients with HER2-positive metastatic breast cancer, including those with brain metastases.
HER2CLIMB is an international, randomized, double-blind trial in which the combination of TUKYSA® plus HERCEPTIN® and XELODA® was compared with placebo plus HERCEPTIN® and XELODA®. A total of 612 patients with unresectable locally advanced or metastatic HER2-positive breast cancer, who were previously treated with HERCEPTIN®, PERJETA® (Pertuzumab) and KADCYLA® (ado-Trastuzumab emtansine) were enrolled. Patients were randomly assigned in a 2:1 ratio to receive either TUKYSA® 300 mg orally twice daily throughout the treatment period (N=410) or placebo orally twice daily (N=201), in combination with HERCEPTIN® 6 mg/kg IV once every 21 days, following an initial loading dose of 8 mg/kg, and XELODA® 1000 mg/m2 orally twice daily on days 1 to 14 of each 21-day cycle. Stratification factors included presence or absence of brain metastases, ECOG Performance Status and geographic region. The median patient age was 54 years and patient demographic as well as disease characteristics at baseline were well balanced between the two treatment groups. In the total treatment population, 47.5% had brain metastases at baseline, 48.3% in the TUKYSA® combination group and 46% in the placebo combination group. The median duration of follow up in the total treatment population was 14 months. The Primary endpoint was Progression Free Survival (PFS) among the first 480 patients who underwent randomization. Secondary end points assessed in the total treatment population (612 patients) included, Overall Survival (OS), PFS among patients with brain metastases, confirmed Objective Response Rate (ORR), and safety.
The Primary endpoint of PFS at 1 year was 33.1% in the TUKYSA®-combination group and 12.3% in the placebo-combination group (HR for disease progression or death=0.54; P<0.001), and the median duration of PFS was 7.8 months and 5.6 months, respectively. This represented a 46% reduction in the risk of cancer progression or death in the TUKYSA®-combination group compared to patients who received HERCEPTIN® and XELODA® alone. The Overall Survival at 2 years was 44.9% in the TUKYSA®-combination group and 26.6% in the placebo-combination group (HR for death=0.66; P=0.005), and the median Overall Survival was 21.9 months and 17.4 months, respectively. This represented a 44% reduction in the risk of death in the TUKYSA®-combination group compared to the placebo-combination group. Among the patients with brain metastases, PFS at 1 year was 24.9% in the TUKYSA®-combination group and 0% in the placebo-combination group (HR=0.48; P<0.001), and the median PFS was 7.6 months and 5.4 months, respectively. This represented a 52% reduction in the risk of cancer progression or death in the TUKYSA®-combination group compared to the placebo-combination group. Among the patients with measurable disease at baseline, the confirmed Objective Response Rate was 40.6% in the TUKYSA®-combination group and 22.8% in the placebo-combination group (P<0.001). Common adverse events in the TUKYSA® group included diarrhea, Palmar-Plantar Erythrodysesthesia syndrome, nausea, vomiting and fatigue. Diarrhea and abnormal liver function tests were more common in the TUKYSA®-combination group than in the placebo-combination group.
It was concluded that in heavily pretreated patients with HER2-positive metastatic breast cancer, including those with brain metastases, the addition of TUKYSA® to HERCEPTIN® and XELODA® resulted in clinically significant improvement in PFS and OS, compared to the placebo-combination group. This trial is unique in that it included patients with active brain metastases, either untreated or progressing.
Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. Murthy RK, Loi S, Okines A, et al. N Engl J Med 2020;382:597-609.
FDA Approves PEMAZYRE®, First Targeted Therapy for Cholangiocarcinoma
SUMMARY: The FDA on April 17, 2020 granted accelerated approval to PEMAZYRE® (Pemigatinib), for the treatment of adults with previously treated, unresectable locally advanced or metastatic Cholangiocarcinoma with a Fibroblast Growth Factor Receptor 2 (FGFR2) fusion or other rearrangement, as detected by an FDA-approved test. The FDA also approved the FoundationOne® CDX (Foundation Medicine, Inc.), as a companion diagnostic for patient selection.
Bile Duct cancer (Cholangiocarcinoma), comprise about 30% of all primary liver tumors and includes both intrahepatic and extrahepatic bile duct cancers. Klatskin tumor is a type of Cholangiocarcinoma that begins in the hilum, at the junction of the left and right bile ducts. It is the most common type of Cholangiocarcinoma, accounting for more than half of all cases. About 8,000 people in the US are diagnosed with Cholangiocarcinoma each year and approximately 20% of the cases are suitable for surgical resection, whereas a majority of patients at diagnosis have advanced disease. The 5-year survival is less than 10%, with limited progress made over the past two decades. There is therefore an unmet need for new effective therapies.
FGFRs (Fibroblast Growth Factor Receptors) play an important role in tumor cell proliferation and survival, migration and angiogenesis. Activating fusions, rearrangements, translocations and gene amplifications in FGFRs result in dysregulation of FGFR signaling, and may contribute to the pathogenesis of various cancers, including Cholangiocarcinoma. FGFR2 fusions or rearrangements occur almost exclusively in intrahepatic Cholangiocarcinoma, where they are observed in 10-16% of patients. PEMAZYRE® is a potent, selective, oral kinase inhibitor of FGFR isoforms 1, 2 and 3, which in preclinical studies has demonstrated selective pharmacologic activity against cancer cells with FGFR alterations.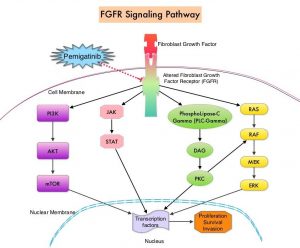
The FIGHT-202 ((FIbroblast Growth factor receptor in oncology and Hematology Trials) is a Phase II, multi-center, open-label, single-arm study which evaluated the safety and efficacy of PEMAZYRE® (Pemigatinib) in adult patients with previously treated, locally advanced or metastatic Cholangiocarcinoma with documented FGFR2 fusion or rearrangement. Patients were enrolled into one of three cohorts: Cohort A with FGFR2 fusions or rearrangements (N=107), Cohort B with other FGF/FGFR genetic alterations (N=20) or Cohort C with no FGF/FGFR genetic alterations (N=18). All patients received PEMAZYRE® 13.5 mg orally once daily, two weeks on and one week off, on a 21-day cycle, until radiological disease progression or unacceptable toxicity. The median patient age of the entire enrolled patient group was 59 years and patients were scanned every eight weeks to assess response to PEMAZYRE®. The Primary endpoint of FIGHT-202 was Objective Response Rate (ORR) in Cohort A, assessed by Independent Review per RECIST criteria. Secondary endpoints included ORR in Cohorts B, A plus B, and C, Duration of Response (DOR), Disease Control Rate (DCR), Progression Free Survival (PFS), Overall Survival (OS), and safety.
It was noted that PEMAZYRE® monotherapy resulted in an Objective Response Rate of 36%, with Complete Response Rate of 2.8% and a Partial Response Rate of 33%. Among those who had a response, 63% had a response lasting 6 months or longer and 18% had a response lasting 12 months or longer. The median Duration of Response was 7.5 months and the Disease Control Rate (DCR) was 82%. The median PFS and median OS were 6.9 months and 21.1 months, and the OS data was not mature at the time of data cutoff. In Cohorts B and C, none of the patients achieved a response. The most common Adverse Events were hyperphosphatemia, alopecia, diarrhea, fatigue, nail toxicities and dysgeusia. Hyperphosphatemia was managed with diet modifications, phosphate binders, diuretics or dose modifications. Fewer patients discontinued therapy in Cohort A compared to Cohort B and C.
It was concluded that based on Overall Response Rate and Duration of Response, PEMAZYRE® is the first and only FDA-approved treatment for previously treated patients with Cholangiocarcinoma, harboring FGFR2 gene rearrangements or fusions.
Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Abou-Alfa GK, Sahai V, Hollebecque A, et al. Lancet Oncol. 2020 Mar 20. pii: S1470-2045(20)30109-1. doi: 10.1016/S1470-2045(20)30109-1. [Epub ahead of print]
JELMYTO® (Mitomycin)
The FDA on April 15, 2020 approved JELMYTO® for adult patients with low-grade upper tract urothelial cancer. JELMYTO® is a product of UroGen Pharma.
TUKYSA® (Tucatinib)
The FDA on April 17, 2020 approved TUKYSA® in combination with Trastuzumab and XELODA® (Capecitabine), for adult patients with advanced unresectable or metastatic HER2-positive breast cancer, including patients with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting. TUKYSA® is a product of Seattle Genetics, Inc.
FDA Approves BRAFTOVI® in Combination with ERBITUX® for Metastatic Colorectal Cancer
SUMMARY: The FDA on April 8, 2020, approved BRAFTOVI® (Encorafenib) in combination with ERBITUX® (Cetuximab) for the treatment of adult patients with metastatic ColoRectal Cancer (CRC) with a BRAF V600E mutation, detected by an FDA-approved test, after prior therapy. Colorectal Cancer is the third most common cancer diagnosed in both men and women in the United States. The American Cancer Society estimates that approximately 147,950 new cases of CRC will be diagnosed in the United States in 2020 and about 53,200 patients are expected to die of the disease. The lifetime risk of developing CRC is about 1 in 23.
Advanced colon cancer is often incurable and standard chemotherapy when combined with anti EGFR (Epidermal Growth Factor Receptor) targeted monoclonal antibodies such as VECTIBIX® (Panitumumab) and ERBITUX® (Cetuximab) as well as anti VEGF agent AVASTIN® (Bevacizumab), have demonstrated improvement in Progression Free Survival (PFS) and Overall Survival (OS). The benefit with anti EGFR agents however is only demonstrable in patients with metastatic CRC (mCRC), whose tumors do not harbor KRAS mutations in codons 12 and 13 of exon 2 (KRAS Wild Type). It is now also clear that even among the KRAS Wild Type patient group about 15-20% have other rare mutations such as NRAS and BRAF mutations, which confer resistance to anti EGFR agents. Patients with stage IV colorectal cancer are now routinely analyzed for extended RAS and BRAF mutations. KRAS mutations are predictive of resistance to EGFR targeted therapy. Approximately 8-15% of all metastatic CRC tumors present with BRAF V600E mutations and BRAF V600E is recognized as a marker of poor prognosis in this patient group. These patients tend to have aggressive disease with a higher rate of peritoneal metastasis and do not respond well to standard treatment intervention. Approximately 20% of the BRAF-mutated population in the metastatic setting has MSI-High tumors, but MSI-High status does not confer protection to this patient group.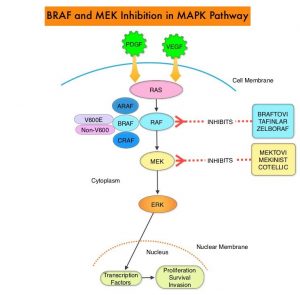
The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. The BRAF V600E mutations results in constitutive activation of the MAP kinase pathway. Inhibiting BRAF can transiently reduce MAP kinase signaling. However, this can result in feedback upregulation of EGFR signaling pathway, which can then reactivate the MAP kinase pathway. This aberrant signaling can be blocked by dual inhibition of both BRAF and EGFR. It should be noted that BRAF V600E-mutated CRC is inherently less sensitive to BRAF inhibition than Malignant Melanoma.
BRAFTOVI® (Encorafenib) is a BRAF inhibitor and has target binding characteristics that differ from other BRAF inhibitors such as ZELBORAF® (Vemurafenib) and TAFINLAR® (Dabrafenib), with a prolonged target dissociation half-life and higher potency. The combination of BRAFTOVI® along with anti-EGFR monoclonal antibody ERBITUX® (Cetuximab) showed promising activity in early-phase clinical trials.
The present FDA approval was based on BEACON CRC (Binimetinib, Encorafenib, and Cetuximab Combined to Treat BRAF-Mutant Colorectal Cancer) trial, which is an international, multicenter, randomized, open-label, Phase III study in which the efficacy and safety of BRAFTOVI® plus ERBITUX® with or without a MEK inhibitor MEKTOVI® (Binimetinib), was compared with the investigators’ choice of ERBITUX® combined with either Irinotecan or Fluorouracil, Folinic acid, and Irinotecan, in patients with BRAF V600E-mutant mCRC, whose disease has progressed after one or two prior regimens. Eligible patients were required to have BRAF V600E mutation-positive metastatic CRC (detected by the Qiagen therascreen® BRAF V600E RGQ PCR kit), with disease progression after one or two prior regimens. In this trial, 665 patients were randomly assigned in a 1:1:1 ratio to receive either triplet therapy of BRAFTOVI® 300 mg orally daily, MEKTOVI® 45 mg orally twice daily, and ERBITUX® 400 mg/m2 IV as an initial dose, then 250 mg/m2 IV weekly (N=224), doublet-therapy of BRAFTOVI® and ERBITUX® administered in the same doses and on the same schedule as the triplet regimen (N=220) or investigators’ choice of ERBITUX® combined with either Irinotecan or Fluorouracil, Folinic acid, and Irinotecan (N=221). Patients were stratified according to previous Irinotecan use and treatment was administered in 28-day cycles until disease progression. The co-Primary end points were Overall Survival (OS) in the triplet-therapy group as compared with the control group and Secondary end points included OS in the doublet-therapy group as compared with the control group, as well as Progression Free Survival, Duration of Response, and Safety in all groups. This study was not powered to compare the triplet-therapy group against the doublet-therapy group. The Overall Response Rate (ORR) and Duration of Response were assessed by blinded Independent Central Review in the subset of the first 220 patients assigned to receive either BRAFTOVI® and ERBITUX® or the control group.
The median OS was 8.4 months in the BRAFTOVI® plus ERBITUX® group, compared to 5.4 months in the control group (HR=0.60; P=0.0003), and this represented 40% reduction in the risk of death among the BRAFTOVI® plus ERBITUX® group. Median PFS was 4.2 months in the BRAFTOVI® plus ERBITUX® group compared to 1.5 months in the control group (HR=0.40; P< 0.0001). The ORR was 20% and 2% respectively. The median Duration of Response was 6.1 months for the BRAFTOVI® plus ERBITUX® group and Not Reached in the control arm. The median OS was 9.0 months in the triplet-therapy group and 5.4 months in the control group (HR for death=0.52; P<0.001). This represented 48% reduction in the risk of death in the triplet-therapy group. Both the triplet and doublet regimens reduced the risk of Quality of Life (QoL) deterioration by about 45% by different QoL assessment instruments, compared with the control regimen. The most common adverse reactions in the BRAFTOVI® plus ERBITUX® group were fatigue, nausea, diarrhea, dermatitis acneiform, abdominal pain, decreased appetite, arthralgia, and rash.
It was concluded from the BEACON CRC trial that a combination of BRAFTOVI®, MEKTOVI® and ERBITUX® as well as a combination of BRAFTOVI® plus ERBITUX® resulted in significantly longer Overall Survival and a higher Response Rate than standard therapy, in patients with metastatic Colorectal Cancer, with the BRAF V600E mutation.
Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. Kopetz S, Grothey A, Yaeger R, et al. N Engl J Med 2019; 381:1632-1643
FDA Approves KEYTRUDA® for BCG-Unresponsive, High-Risk Non-Muscle Invasive Bladder Cancer
SUMMARY: The FDA on January 8, 2020, approved KEYTRUDA® (Pembrolizumab) for the treatment of patients with Bacillus Calmette-Guerin (BCG)-unresponsive, high-risk, Non-Muscle Invasive Bladder Cancer (NMIBC) with Carcinoma In Situ (CIS) with or without papillary tumors, who are ineligible for or have elected not to undergo cystectomy.
The American Cancer Society estimates that for 2020, about 81,400 new cases of bladder cancer will be diagnosed in the US and about 17,980 patients will die of the disease. Bladder cancer is the fourth most common cancer in men, but is less common in women and the average age at the time of diagnosis is 73. Approximately 50% of all bladder cancers are non-invasive or in situ cancers. Patients with high-risk, Non-Muscle Invasive Bladder Cancer that has become unresponsive to BCG treatment, are often given the treatment option of radical cystectomy, which includes removing the entire urinary bladder and a prostatectomy for men or total hysterectomy in women. While highly curative, this surgical procedure carries substantial risk for morbidity and mortality, and can negatively impact patient’s quality of life. Further, a significant proportion of patients are medically ineligible for a radical cystectomy, and even if eligible, refuse surgery and opt for other less effective treatments, which could compromise outcomes.
KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. By doing so, it unleashes the tumor-specific effector T cells, and is thereby able to undo PD-1 pathway-mediated inhibition of the immune response. KEYTRUDA® is presently approved by the FDA for the treatment of patients with locally advanced or metastatic Urothelial carcinoma who are not eligible for Cisplatin-containing chemotherapy or for those with disease progression during or following platinum-containing chemotherapy, based on its durable antitumor activity in this patient group. Upregulation of the PD-1 pathway has been observed in BCG-resistant NMIBC, suggesting that KEYTRUDA® may be of benefit in this group of patients.
This new FDA approval for KEYTRUDA® was based on the KEYNOTE-057 study, which is a multicenter, single-arm trial that enrolled 148 patients with high-risk NMIBC, of whom 96 patients had BCG-unresponsive CIS with or without papillary tumors. BCG-unresponsive high-risk Non-Muscle Invasive Bladder Cancer was defined as persistent disease despite adequate BCG therapy, disease recurrence after an initial tumor-free state following adequate BCG therapy, or T1 disease following a single induction course of BCG. Eligible patients had received adequate BCG therapy and were unable/unwilling to undergo radical cystectomy. All patients had undergone TransUrethral Resection of Bladder Tumor (TURBT) to remove resectable disease. Patients with residual Carcinoma In Situ, not amenable to complete resection were permitted. Patients received KEYTRUDA® 200 mg every 3 weeks until unacceptable toxicity, persistent or recurrent high-risk NMIBC or progressive disease, or up to 24 months of therapy without disease progression. The median age was 73 years and the median number of prior BCG instillations was 12. More than half of patients (56.9%) had a PD-L1 Combined Positive Score (CPS) of less than 10, and most patients in this analysis had refused prior cystectomy. The Primary end point was Complete Response Rate (CRR) as defined by negative results for cystoscopy with TURBT/biopsies as applicable, urine cytology, and CT Urography imaging. Secondary end points included Duration of Response and Safety.
At a median follow up was 28 months the Complete Response Rate was 41% and the median Duration of Response was 16.2 months. Forty-six percent (46%) of responding patients experienced a Complete Response lasting at least 12 months. The most frequent adverse reactions were fatigue, diarrhea, rash, pruritis, musculoskeletal pain, peripheral edema and hypothyroidism.
It was concluded from this study that KEYTRUDA® had encouraging activity in bladder cancer patients, with high-risk, BCG-unresponsive Carcinoma in Situ, with or without papillary tumors. The authors added that this study demonstrates that immune activation with systemically administered treatment can result in local activity in the bladder, as well as long-term durable remissions of cancer.
Keynote 057: Phase II trial of Pembrolizumab (pembro) for patients (pts) with high-risk (HR) nonmuscle invasive bladder cancer (NMIBC) unresponsive to bacillus calmette-guérin (BCG). Balar AV, Kulkarni GS, Uchio EM, et al. J Clin Oncol 37, 2019 (suppl 7S; abstr 350)
BRAFTOVI® (Encorafenib) and ERBITUX® (Cetuximab)
The FDA on April 8, 2020 approved BRAFTOVI® (Encorafenib), in combination with ERBITUX® (Cetuximab) for the treatment of adult patients with metastatic ColoRectal Cancer (CRC) with a BRAF V600E mutation, detected by an FDA-approved test, after prior therapy. BRAFTOVI® is a product of Array BioPharma Inc.
REBLOZYL® (Luspatercept-aamt)
The FDA on April 3, 2020, approved REBLOZYL® (Luspatercept-aamt) for the treatment of anemia, failing an Erythropoiesis Stimulating Agent, and requiring 2 or more RBC units over 8 weeks, in adult patients with very low- to intermediate-risk MyeloDysplastic Syndromes with Ring Sideroblasts (MDS-RS), or with MyeloDysplastic/MyeloProliferative Neoplasm with Ring Sideroblasts and Thrombocytosis (MDS/MPN-RS-T). REBLOZYL® is a product of Celgene Corporation.