SUMMARY: The FDA on March 27, 2020 approved IMFINZI® (Durvalumab) in combination with Etoposide and either Carboplatin or Cisplatin as first-line treatment for patients with Extensive-Stage Small Cell Lung Cancer (ES-SCLC). The American Cancer Society estimates that for 2020 about 228,820 new cases of lung cancer will be diagnosed and about 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Small cell lung cancer (SCLC) accounts for approximately 13-15% of all lung cancers and is aggressive. Patients with extensive stage SCLC are often treated with a combination of Carboplatin or Cisplatin with Etoposide as first line treatment and the tumor response rates are as high as 60-80%. However, majority of the patients relapse within months of completing initial therapy, with a median Overall Survival (OS) of approximately 10 months. Patients often receive HYCAMTIN® (Topotecan) for recurrent or progressive SCLC (second-line treatment) and after failure on second-line therapy, treatment options are limited. The 5 year survival rate for Extensive Stage SCLC (ES-SCLC) is less than 5%, with a median survival of 9-10 months from the time of diagnosis.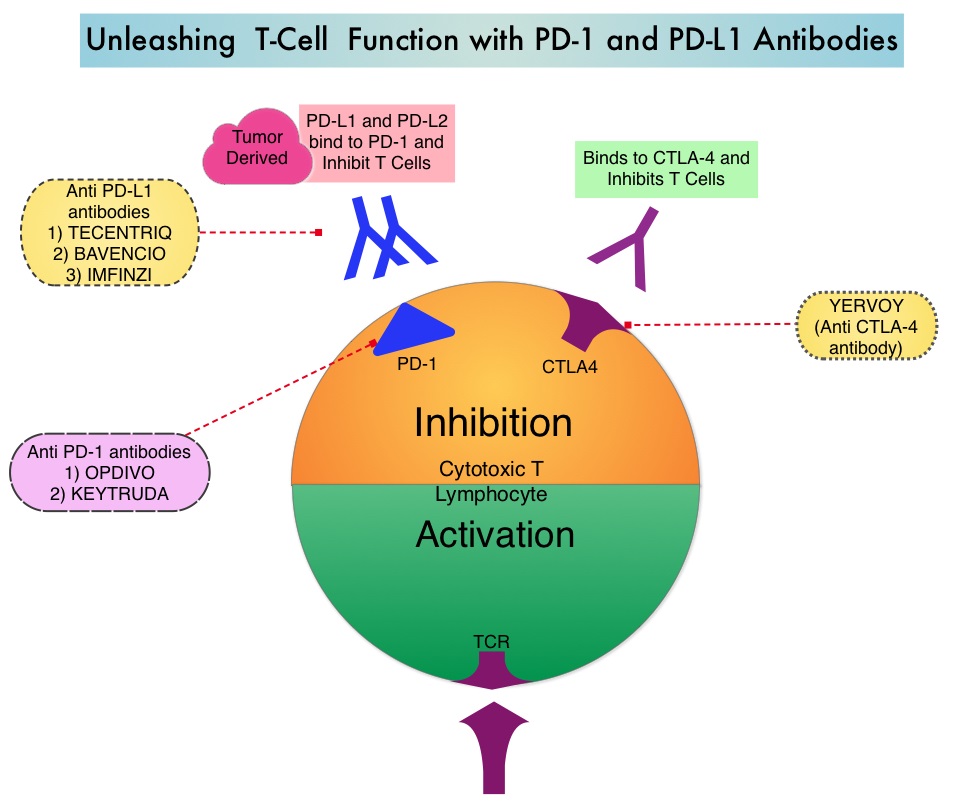
Based on the premise that SCLC has a high mutation rate, it was hypothesized that these tumors may be immunogenic and more recently immunotherapy with checkpoint inhibitors has demonstrated clinical activity in Extensive Stage SCLC. IMFINZI® (Durvalumab) is a selective, high-affinity, human IgG1 monoclonal antibody, that blocks the binding of Programmed Death Ligand 1 (PD-L1) to Programmed Death 1 (PD-1) receptor and CD80, thereby unleashing the T cells to recognize and kill tumor cells.
IMFINZI® is approved by the FDA for the treatment of patients with locally advanced, unresectable Stage III Non-Small Cell Lung Cancer, who have not progressed following chemoradiotherapy. Additionally, IMFINZI® is also approved for the treatment of patients with locally advanced or metastatic Urothelial carcinoma who have disease progression during or following Platinum-containing chemotherapy, or who have disease progression within 12 months of neoadjuvant or adjuvant treatment with Platinum-containing chemotherapy.
This present FDA approval was based on CASPIAN trial, which is a multicenter, randomized, controlled, open-label, Phase III study, in which the efficacy of IMFINZI® with or without CTLA-4 inhibitor Tremelimumab, in combination with Etoposide plus either Cisplatin or Carboplatin (Platinum-Etoposide), was compared to chemotherapy alone, in treatment-naive patients with ES-SCLC. Patients were randomly assigned in a 1:1:1 ratio to IMFINZI® plus Platinum-Etoposide, IMFINZI® plus Tremelimumab plus Platinum-Etoposide, or Platinum-Etoposide alone. This study allocated 268 patients to the IMFINZI® plus Platinum-Etoposide group and 269 patients to the Platinum-Etoposide group. Treatment with Platinum-Etoposide consisted of Etoposide 80-100 mg/m2 IV on days 1-3 of each cycle with investigator’s choice of either Carboplatin AUC 5-6 mg/mL per min or Cisplatin 75-80 mg/m2 IV administered on day 1 of each cycle. Patients received up to four cycles of Platinum-Etoposide along with IMFINZI® 1500 mg IV with or without Tremelimumab 75 mg IV every 3 weeks, followed by maintenance IMFINZI® 1500 mg IV every 4 weeks in the immunotherapy treatment groups, or up to six cycles of Platinum-Etoposide IV every 3 weeks plus Prophylactic Cranial Irradiation (at the treating physicians discretion), in the Platinum-Etoposide control group. The median patient age was 62 years and 10% of the patients had CNS metastases. PCI was administered to 8% of patients in the Platinum-Etoposide group. The Primary endpoint was Overall Survival (OS). Additional efficacy outcome measures included Progression Free Survival (PFS) and Objective Response Rate (ORR). The authors reported the results for the IMFINZI® plus Platinum-Etoposide group, compared to the Platinum-Etoposide group, from a planned interim analysis.
The median OS was 13.0 months in the IMFINZI® plus chemotherapy group, compared with 10.3 months in the chemotherapy alone group (HR=0.73; P=0.0047), with a 27% reduction in the risk of death. Approximately 34% of patients who received IMFINZI® were alive at 18 months as compared to 25% in the control arm of the trial. Additionally, IMFINZI® plus chemotherapy demonstrated a higher PFS rate at 12 months (17.5% versus 4.7%), a 10.3% increase in confirmed ORR (67.9% versus 57.6%), and improved Duration of Response at 12 months (22.7% versus 6.3%). The most common adverse reactions noted were nausea, fatigue, asthenia, and alopecia.
It was concluded that the addition of IMFINZI® to first line Platinum-Etoposide chemotherapy combination significantly improved Overall Survival in patients with Extensive Stage-Small Cell Lung Cancer, when compared to chemotherapy alone.
Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Paz-Ares L, Dvorkin M, Chen Y, CASPIAN investigators, et al. Lancet. 2019;394:1929-1939