
The FDA on May 8, 2020, expanded the indication of LYNPARZA® to include its combination with Bevacizumab for first-line maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in Complete or Partial Response to first-line Platinum-based chemotherapy, and whose cancer is associated with Homologous Recombination Deficiency (HRD) positive status, defined by either a deleterious or suspected deleterious BRCA mutation, and/or genomic instability. LYNPARZA® is a product of AstraZeneca Pharmaceuticals, LP.
Tag: Ovarian Cancer
FDA Approves ZEJULA® for Newly Diagnosed Patients with Advanced Ovarian Cancer
SUMMARY: The FDA on April 29, 2020 approved ZEJULA® (Niraparib) for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a complete or partial response to first-line platinum-based chemotherapy. It is estimated that in the United States, approximately 21,750 women will be diagnosed with ovarian cancer in 2020 and 13,940 women will die of the disease. Ovarian cancer ranks fifth in cancer deaths among women, and accounts for more deaths than any other cancer of the female reproductive system. Approximately 75% of the ovarian cancer patients are diagnosed with advanced disease. Patients with newly diagnosed advanced ovarian cancer are often treated with platinum based chemotherapy following primary surgical cytoreduction. Approximately 70% of these patients will relapse within the subsequent 3 years and are incurable, with a 5 year Overall Survival rate of about 20-30%.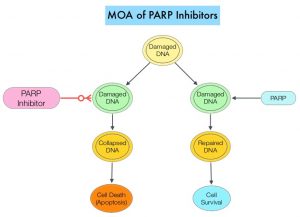
DNA damage is a common occurrence in daily life by UV light, ionizing radiation, replication errors, chemical agents, etc. This can result in single and double strand breaks in the DNA structure which must be repaired for cell survival. The two vital pathways for DNA repair in a normal cell are BRCA1/BRCA2 and PARP. The PARP (Poly ADP Ribose Polymerase) family of enzymes, include PARP1 and PARP2. In the context of DNA repair, BRCA1 and BRCA2 genes recognize and repair double strand DNA breaks via Homologous Recombination (HR) pathway. Homologous Recombination is a type of genetic recombination, and is a DNA repair pathway utilized by cells to accurately repair DNA double-stranded breaks during the S and G2 phases of the cell cycle, and thereby maintain genomic integrity. Homologous Recombination Deficiency (HRD) is noted following mutation of genes involved in HR repair pathway. At least 15 genes are involved in the Homologous Recombination Repair (HRR) pathway including BRCA1 and BRCA2 genes. The BRCA1 gene is located on the long (q) arm of chromosome 17 whereas BRCA2 is located on the long arm of chromosome 13. BRCA1 and BRCA2 are tumor suppressor genes and functional BRCA proteins repair damaged DNA, and play an important role in maintaining cellular genetic integrity. They regulate cell growth and prevent abnormal cell division and development of malignancy. Mutations in BRCA1 and BRCA2 account for about 20-25% of hereditary breast cancers and about 5-10% of all breast cancers. They also account for 15% of ovarian cancers, in addition to other cancers such as Colon and Prostate. BRCA mutations can either be inherited (Germline) and present in all individual cells or can be acquired and occur exclusively in the tumor cells (Somatic). Somatic mutations account for a significant portion of overall BRCA1 and BRCA2 aberrations. Loss of BRCA function due to frequent somatic aberrations likely deregulates HR pathway, and other pathways then come in to play, which are less precise and error prone, resulting in the accumulation of additional mutations and chromosomal instability in the cell, with subsequent malignant transformation. HRD therefore indicates an important loss of DNA repair function. Hereditary Epithelial Ovarian Cancer was thought to be caused almost exclusively by mutations in BRCA1 and BRCA2. It however is now well known that about 50% of the high grade serous ovarian cancers have aberrations in HR repair pathway. Deregulated HR pathway increases sensitivity to platinum drugs. Majority of the women with germline BRCA mutations (gBRCA) are positive for HR deficiency.
PARP is a related enzymatic pathway that repairs single strand breaks in DNA. In a BRCA mutant, the cancer cell relies solely on PARP pathway for DNA repair. In the presence of a PARP inhibitor, there is synthetic lethality because loss of both genes, leading to cell death. Thus PARP inhibitors are only harmful to cancer cells. ZEJULA® is a highly selective PARP 1/2 inhibitor, that causes cumulative DNA damage and cell death by inhibiting PARP. Previously published phase III study among patients with platinum-sensitive, recurrent ovarian cancer (NEJM 2016;375:2154-2164) concluded that Niraparib significantly prolonged Progression Free Survival (PFS) compared to placebo, and this benefit was achieved regardless of the presence or absence of germline BRCA mutations or HRD status.
PRIMA trial is a randomized, double-blind, placebo-controlled, international Phase III trial conducted to test the efficacy and safety of ZEJULA® maintenance therapy after a response to platinum-based chemotherapy, in patients with newly diagnosed advanced ovarian cancer at high risk for relapse. It should be noted that at the time PRIMA trial was designed, AVASTIN® (Bevacizumab) was not approved for first-line treatment in all participating countries. A total of 733 patients with newly diagnosed, high risk, advanced ovarian cancer were randomly assigned in a 2:1 ratio to receive ZEJULA® (N=487) or placebo (N=246) once daily in 28-day cycles for 36 months or until disease progression, after a response to platinum-based chemotherapy regimen. Patients received a dose of 200-300mg once daily, based on body weight and platelet count. Enrolled patients were at high risk for progressive disease with 23.1% having Stage III ovarian cancer with residual disease after primary debulking surgery, 66.7% had received neoadjuvant chemotherapy, 35% had Stage IV ovarian cancer, and 30.5% had a Partial Response to first-line platinum-based chemotherapy. Tumor samples were tested for HRD status and HRD was defined by either presence of tumor BRCA mutation or Genomic Instability Score (GIS) of 42 or more. Of the randomized patients, 50.9% had tumors with HRD, 30.4% had BRCA mutations and 20.5% were BRCA wild type. The treatment groups were well balanced. The Primary endpoint was Progression Free Survival (PFS) in patients who had tumors with HRD, and then in the overall population, as determined on hierarchical testing. Secondary end points included Overall Survival, time until the first subsequent therapy, PFS 2, defined as time from randomization to progression while the patient was receiving a subsequent anticancer therapy and Patient-Reported Outcomes. The median duration of follow-up at the time of the data cutoff was 13.8 months.
There was a statistically significant improvement in PFS for patients randomized to ZEJULA® compared with placebo in the HRD group, as well as the overall population. The median PFS in the HRD group was 21.9 months for patients receiving ZEJULA® compared with 10.4 months for those receiving placebo (HR=0.43; P<0.001). The median PFS in the overall population was 13.8 months for patients receiving ZEJULA® compared with 8.2 months for those receiving placebo (HR=0.62; P<0.001). At the 24-month interim analysis, the rate of Overall Survival was 84% in the ZEJULA® group and 77% in the placebo group (HR=0.70). The most common adverse reactions in patients receiving ZEJULA® were cytopenias, fatigue, AST/ALT elevation, hypertension, low grade nausea and decreased appetite.
It was concluded that among patients with newly diagnosed advanced ovarian cancer who had responded to platinum-based chemotherapy, ZEJULA® significantly prolonged Progression Free Survival, compared to those who received placebo, regardless of the presence or absence of Homologous Recombination Deficiency.
Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. González-Martín A, Pothuri B, Vergote I, et al. for the PRIMA/ENGOT-OV26/GOG-3012 Investigators. N Engl J Med 2019; 381:2391-2402
ZEJULA® (Niraparib)
The FDA on October 23, 2019 approved ZEJULA® for patients with advanced Ovarian, Fallopian tube, or Primary Peritoneal cancer treated with three or more prior chemotherapy regimens and whose cancer is associated with Homologous Recombination Deficiency (HRD)-positive status. HDR is defined by either a deleterious or suspected deleterious BRCA mutation, or genomic instability in patients with disease progression greater than six months after response to the last Platinum-based chemotherapy. ZEJULA® is a product of Tesaro, Inc.
Substantial Benefit with Maintenance LYNPARZA® in Patients with Newly Diagnosed Advanced Ovarian Cancer
SUMMARY: It is estimated that in the United States, approximately 22,530 women will be diagnosed with ovarian cancer in 2019 and 13,980 women will die of the disease. Ovarian cancer ranks fifth in cancer deaths among women, and accounts for more deaths than any other cancer of the female reproductive system. Approximately 75% of the ovarian cancer patients are diagnosed with advanced disease. Patients with newly diagnosed advanced ovarian cancer are often treated with platinum based chemotherapy following primary surgical cytoreduction. Approximately 70% of these patients will relapse within the subsequent 3 years and are incurable, with a 5 year Overall Survival rate of about 20-30%. 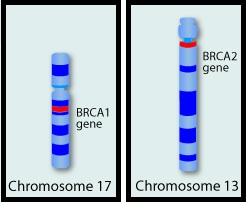
BRCA1 and BRCA2 are tumor suppressor genes and functional BRCA proteins that repair damaged DNA, and play an important role in maintaining cellular genetic integrity. They regulate cell growth and prevent abnormal cell division and development of malignancy. Mutations in BRCA1 and BRCA2 account for about 20-25% of hereditary breast cancers and about 5-10% of all breast cancers. They also account for 15% of ovarian cancers, in addition to other cancers such as colon and prostate. BRCA mutations can either be inherited (Germline) and present in all individual cells or can be acquired and occur exclusively in the tumor cells (Somatic). Somatic mutations account for a significant portion of overall BRCA1 and BRCA2 aberrations. Loss of BRCA function due to frequent somatic aberrations in ovarian cancers likely deregulates Homologous Recombination (HR) pathway and increases sensitivity to platinum drugs. Majority of the women with Germline BRCA mutations (gBRCA) are positive for HR deficiency. The PARP (Poly ADP Ribose Polymerase) family of enzymes which include PARP1 and PARP2, repair damaged DNA. PARP inhibitors kill tumors defective in the BRCA1 or BRCA2 genes through the concept of synthetic lethality. Epithelial ovarian cancers with Homologous Recombination Deficiency have demonstrated sensitivity to PARP inhibitors.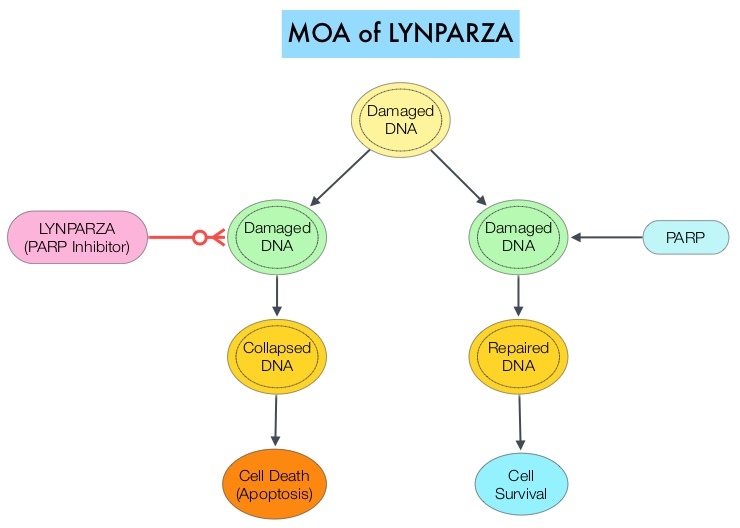
SOLO1 is an international, randomized, double-blind, Phase III trial, conducted to evaluate the efficacy of maintenance therapy with a PARP inhibitor LYNPARZA® (Olaparib), in patients with newly diagnosed advanced ovarian cancer with a Germline or Somatic mutation in BRCA1, BRCA2, or both (BRCA1/2), who had a complete or partial clinical response after platinum-based chemotherapy. Patients (N=391) were randomly assigned in a 2:1 ratio, to receive LYNPARZA® tablets 300 mg PO twice daily (N=260) or placebo (N=131). Enrolled patients had international FIGO Stage III or IV high-grade Serous or Endometrioid ovarian cancer, Primary Peritoneal cancer, or Fallopian tube cancer (or a combination thereof), and majority of the enrolled patients (N=388) had a centrally confirmed Germline BRCA1/2 mutation, and 2 patients had a centrally confirmed Somatic BRCA1/2 mutation. Patients with Stage III disease had cytoreductive surgery attempted upfront before the start chemotherapy, or had interval cytoreductive surgery after the start but before the end of chemotherapy. Patients with Stage IV disease either had a biopsy for tissue diagnosis or underwent upfront or interval cytoreductive surgery. The Primary end point was Progression Free Survival (PFS) and Secondary end points included second PFS which was the time from randomization to second disease progression or death, and Overall Survival.
After a median follow up of 41 months, the risk of disease progression or death was 70% lower with LYNPARZA® when compared to placebo, with an estimated rate of freedom from disease progression and death at 3 years of 60% versus 27% (HR for disease progression or death= 0.30; P<0.001). The estimated rate of freedom from second disease progression and death at 3 years was 75% in the LYNPARZA® group, as compared with 60% in the placebo group (HR for second disease progression or death=0.50; P<0.001). Adverse events were consistent with the known toxic effects of LYNPARZA®, with anemia being the most common serious side effect. Adverse events were usually managed by dose interruption or dose reduction, rather than discontinuation of the study drug.
It was concluded that maintenance therapy with LYNPARZA® after platinum-based chemotherapy provided a substantial Progression Free Survival benefit, with a 70% lower risk of disease progression or death, when compared to placebo, among women with newly diagnosed advanced ovarian cancer and a BRCA1/2mutation. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. Moore K, Colombo N, Scambia G, et al. N Engl J Med 2018; 379:2495-2505
Aspirin Lowers Risk for Ovarian and Hepatocellular Carcinoma
SUMMARY: Aspirin (AcetylSalicylic Acid) has been studied as a chemopreventive agent for several decades and the temporal relationship between systemic inflammation and cancer has been a topic of ongoing investigation. The US Preventive Services Task Force (USPSTF) found adequate evidence that Aspirin use reduces the incidence of ColoRectal Cancer (CRC) in adults after 5-10 years of use, and recommends initiating low-dose Aspirin use for the primary prevention of CardioVascular Disease (CVD) and ColoRectal Cancer (CRC) in adults aged 50-69 years, who have a 10% or greater 10-year CVD risk, are not at increased risk for bleeding, have a life expectancy of at least 10 years, and are willing to take low-dose Aspirin daily for at least 10 years.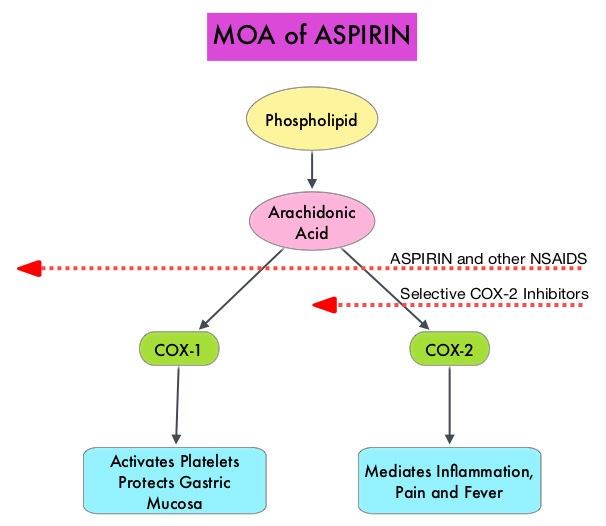
The molecular mechanisms underlying Aspirin’s chemoprevention effects as well as the dose, duration, and timing of Aspirin chemoprevention have remained unclear. More recent data suggests that platelets may play a role in tumorigenesis as well, through the release of angiogenic and growth factors due to overexpression of COX-2. Daily low dose Aspirin inhibits COX-1 and COX-2. It is postulated that Aspirin also works by COX-independent mechanisms such as, the inhibition of NF-kB and Wnt/ β-catenin signaling, which may play a role in its chemopreventive properties.
Two recently published studies examining different doses of Aspirin in different cancers, highlight the beneficial role of Aspirin in reducing cancer risk.
The first report is a large prospective study which attempted to reproduce findings from case-control studies that reported lower Ovarian cancer risk among low-dose Aspirin users. This prospective study evaluated whether regular Aspirin or nonaspirin NonSteroidal Anti-Inflammatory Drug (NSAID) use and patterns of use was associated with lower risk of Ovarian cancer. This cohort study analyzed NSAID use and Ovarian cancer diagnosis data on 205,498 women from 2 prospective cohorts; 93 664 women in the Nurses’ Health Study (NHS), who were 93% non-Hispanic white, with a mean age at baseline of 46 years, followed up from 1980 to 2014, and 111834 women in the Nurses’ Health Study II (NHSII), who were 92% non-Hispanic white, with a mean age at baseline of 34.2 years, followed up from 1989 to 2015. For each analgesic type (Aspirin, low-dose Aspirin, nonaspirin NSAIDs, and Acetaminophen), timing, duration, frequency, and number of tablets used were evaluated, and information was updated every 2-4 years.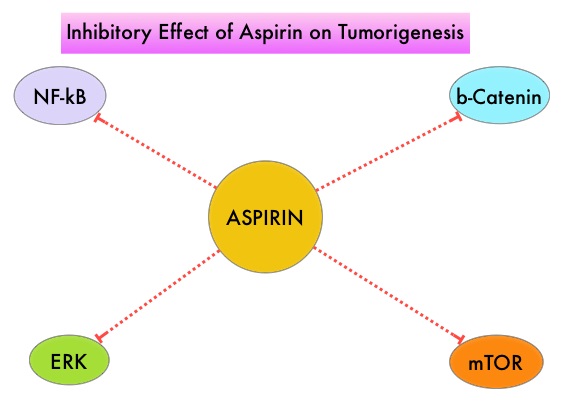
It was noted that among both cohorts, there were 1054 women who developed epithelial Ovarian cancer. Recent use of low-dose Aspirin (100 mg or less) was associated with a lower risk of Ovarian cancer (HR=0.77), whereas there was no such association noted with standard-dose of 325 mg Aspirin (HR=1.17). The associations between Aspirin use and risk of Ovarian cancer did not differ among premenopausal versus postmenopausal women. This study also suggested that use of non-aspirin NSAIDs, such as Ibuprofen and Naproxen, when taken in quantities of at least 10 tablets per week for multiple years was positively associated with an increased risk of Ovarian cancer. There was however no clear associations for the use of Acetaminophen.
The authors concluded that consistent with case-control studies, this prospective analysis showed a reduced risk of Ovarian cancer among regular users of low-dose Aspirin and an increased risk of Ovarian cancer with the use of nonaspirin NSAIDs. These findings suggest that low-dose Aspirin recommended for cardiovascular prophylaxis and ColoRectal Cancer risk reduction can also reduce the risk of Ovarian cancer.
The second report is another large prospective study which analyzed the data on the risk of HepatoCellular Carcinoma (HCC) within 2 populations of a total of 133 371 health care professionals who self reported use of Aspirin. In this pooled analysis, 87 507 were women, with a mean age was 62 years, and 45 864 were men, with a mean age of 64 years. Women reported data biennially since 1980 and men since 1986, on frequency, dosage, and duration of Aspirin use, and data were accessed from November 2017 through March 2018. Individuals with a cancer diagnosis at baseline (except nonmelanoma skin cancer) were excluded.
The researchers noted that regular Aspirin use of 2 or more standard dose 325 mg tablets per week was associated with a 49% reduction in risk of HCC (adjusted HR=0.51) compared to non regular use. This benefit was dose-dependent with the greatest benefit among those taking more than 5 tablets per week (P for trend =0 .006). Further, significantly lower risk for HCC was observed with increasing duration of Aspirin intake (P for trend =0.03), with this decreasing risk noted with the use of 1.5 or more standard-dose Aspirin tablets per week for 5 or more years (adjusted HR=0.41). The use of nonaspirin NSAIDs however was not significantly associated with HCC risk.
The authors from this study concluded that regular and long-term use of standard dose (325 mg) Aspirin, taken at least 2 or more times per week is associated with a dose-dependent reduction in HCC risk, which is apparent after 5 or more years of use.
Taken together, these 2 studies provide the evidence supporting the ability of regular use of Aspirin to prevent Ovarian cancer and HepatoCellular Cancer (HCC). Aspirin is rapidly emerging as a valuable chemoprevention agent for various malignancies.
Association of Analgesic Use With Risk of Ovarian Cancer in the Nurses’ Health Studies. Barnard ME, Poole EM, Curhan GC, et al. JAMA Oncol. 2018;4:1675-1682.
Association Between Aspirin Use and Risk of Hepatocellular Carcinoma. Simon TG, Ma Y, Ludvigsson JF, et al. JAMA Oncol. 2018;4:1683-1690
LYNPARZA® (Olaparib)
The FDA on December 19, 2018 approved LYNPARZA® for the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated (gBRCAm or sBRCAm) advanced epithelial ovarian, fallopian tube or primary peritoneal cancer, who are in Complete or Partial Response to first-line platinum-based chemotherapy. LYNPARZA® is a product of AstraZeneca Pharmaceuticals LP.
AVASTIN® (Bevacizumab)
The FDA on June 13, 2018 approved AVASTIN® for patients with epithelial ovarian, fallopian tube, or primary peritoneal cancer, in combination with Carboplatin and Paclitaxel, followed by single-agent AVASTIN®, for stage III or IV disease, after initial surgical resection. AVASTIN® is a product of Genentech, Inc.
RUBRACA® (Rucaparib)
The FDA on April 6, 2018 approved RUBRACA®, a Poly ADP-Ribose Polymerase (PARP) inhibitor, for the maintenance treatment of recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a Complete or Partial Response to platinum-based chemotherapy. RUBRACA® is a product of Clovis Oncology Inc.
FDA Approves RUBRACA® for Maintenance Treatment of Recurrent Ovarian Cancer
SUMMARY: The FDA on April 6, 2018, approved RUBRACA® (Rucaparib), a Poly ADP-Ribose Polymerase (PARP) inhibitor, for the maintenance treatment of recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a Complete or Partial Response to platinum-based chemotherapy. RUBRACA® was initially approved in December 2016 as monotherapy for the treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer, who have been treated with two or more chemotherapies.
RUBRACA® is an oral, small molecule inhibitor of Poly-Adenosine diphosphate [ADP] Ribose Polymerase (PARP), developed for treatment of ovarian cancer, associated with Homologous Recombination DNA repair deficiency (HRD). Previously published clinical data had suggested that ovarian cancer patients with and without evidence of a germline BRCA mutation, benefit from treatment with a PARP inhibitor, and that maintenance treatment with a PARP inhibitor following a response to platinum-based treatment increases Progression Free Survival (PFS), in patients with ovarian cancer. Even though patients with or without BRCA mutation benefited, the most benefit was derived in those with BRCA mutation.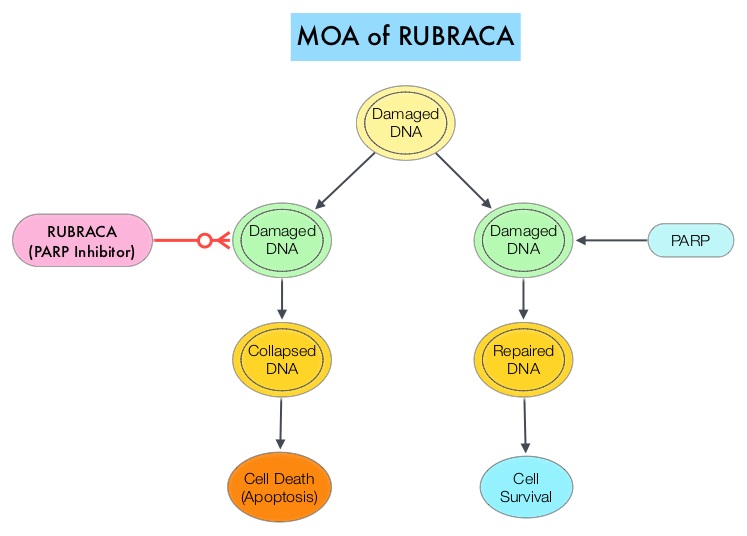
The approval of RUBRACA® was based on ARIEL3, a randomized, double-blind, placebo-controlled, phase III trial, which evaluated the benefit of RUBRACA® versus placebo, after response to second-line or later platinum-based chemotherapy, in patients with high-grade, recurrent, platinum-sensitive ovarian carcinoma. In this trial, 561 patients were randomly assigned in a 2:1 ratio to receive RUBRACA® 600 mg orally twice daily (N=372) or placebo (N=189). Treatment was continued until disease progression or unacceptable toxicity. Eligible patients had recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, and had been treated with at least two prior treatments of platinum-based chemotherapy, and were in Complete or Partial Response to the most recent platinum-based chemotherapy. Patients had CA-125 level of less than the upper limit of normal. Using Next-Generation Sequencing assay, tumor tissue was examined to determine whether DNA contained a deleterious somatic or germline BRCA mutation (tBRCA), in addition to determining the percentage of genomic Loss of Heterozygosity (LOH). Positive Homologous Recombination Deficiency (HRD) status was defined as tBRCA-positive and/or LOH high. The Primary end point was Progression Free Survival in three patient cohorts – all patients, HRD subgroup, and tumor BRCA subgroup.
It was noted that there was a statistically significant improvement in median Progression Free Survival (PFS) for all patients assigned to RUBRACA®, compared with placebo (median PFS 10.8 versus 5.4 months, HR=0.36; P<0.0001). In the HRD subgroup, the median PFS was 13.6 months for those assigned to RUBRACA®, versus 5.4 months for the placebo group (HR=0.32; P<0.0001), and in the tumor BRCA subgroup, the median PFS was 16.6 versus 5.4 months (HR=0.23; P <0.0001), respectively. The most common adverse reactions were fatigue, rash, nausea, vomiting, diarrhea, abdominal discomfort, cytopenias and abnormal liver function studies. Discontinuation due to adverse reactions occurred in 15% of patients receiving RUBRACA®.
It was concluded that RUBRACA® significantly improved Progression Free Survival in patients with platinum-sensitive ovarian cancer who had achieved a response to platinum-based chemotherapy, and could be considered a new standard of care for women with platinum-sensitive ovarian cancer, following a complete or partial response to second-line or later lines of platinum-based chemotherapy. The FDA also concurrently approved the complementary diagnostic test, FoundationFocusTM CDx BRCA LOH for tumor samples, to determine HRD status.
Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Coleman RL, Oza AM, Lorusso D, et al. The Lancet 2017;390:1949-1961