
SUMMARY: The FDA on April 6, 2018, approved RUBRACA® (Rucaparib), a Poly ADP-Ribose Polymerase (PARP) inhibitor, for the maintenance treatment of recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a Complete or Partial Response to platinum-based chemotherapy. RUBRACA® was initially approved in December 2016 as monotherapy for the treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer, who have been treated with two or more chemotherapies.
RUBRACA® is an oral, small molecule inhibitor of Poly-Adenosine diphosphate [ADP] Ribose Polymerase (PARP), developed for treatment of ovarian cancer, associated with Homologous Recombination DNA repair deficiency (HRD). Previously published clinical data had suggested that ovarian cancer patients with and without evidence of a germline BRCA mutation, benefit from treatment with a PARP inhibitor, and that maintenance treatment with a PARP inhibitor following a response to platinum-based treatment increases Progression Free Survival (PFS), in patients with ovarian cancer. Even though patients with or without BRCA mutation benefited, the most benefit was derived in those with BRCA mutation.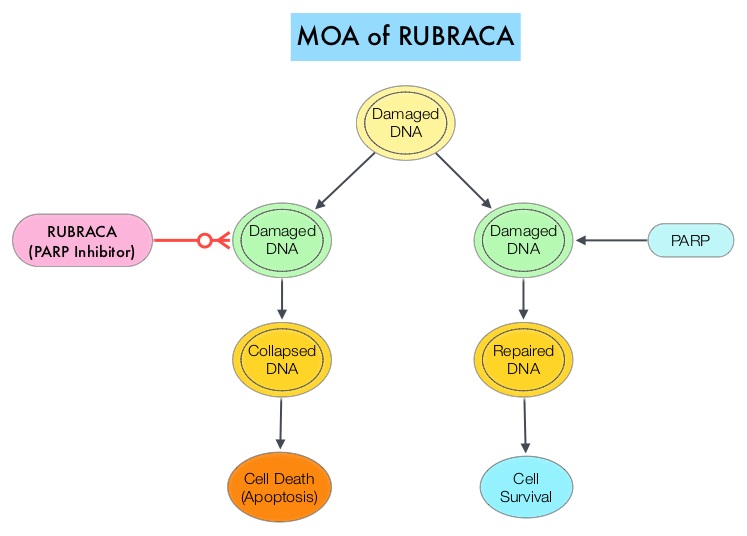
The approval of RUBRACA® was based on ARIEL3, a randomized, double-blind, placebo-controlled, phase III trial, which evaluated the benefit of RUBRACA® versus placebo, after response to second-line or later platinum-based chemotherapy, in patients with high-grade, recurrent, platinum-sensitive ovarian carcinoma. In this trial, 561 patients were randomly assigned in a 2:1 ratio to receive RUBRACA® 600 mg orally twice daily (N=372) or placebo (N=189). Treatment was continued until disease progression or unacceptable toxicity. Eligible patients had recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, and had been treated with at least two prior treatments of platinum-based chemotherapy, and were in Complete or Partial Response to the most recent platinum-based chemotherapy. Patients had CA-125 level of less than the upper limit of normal. Using Next-Generation Sequencing assay, tumor tissue was examined to determine whether DNA contained a deleterious somatic or germline BRCA mutation (tBRCA), in addition to determining the percentage of genomic Loss of Heterozygosity (LOH). Positive Homologous Recombination Deficiency (HRD) status was defined as tBRCA-positive and/or LOH high. The Primary end point was Progression Free Survival in three patient cohorts – all patients, HRD subgroup, and tumor BRCA subgroup.
It was noted that there was a statistically significant improvement in median Progression Free Survival (PFS) for all patients assigned to RUBRACA®, compared with placebo (median PFS 10.8 versus 5.4 months, HR=0.36; P<0.0001). In the HRD subgroup, the median PFS was 13.6 months for those assigned to RUBRACA®, versus 5.4 months for the placebo group (HR=0.32; P<0.0001), and in the tumor BRCA subgroup, the median PFS was 16.6 versus 5.4 months (HR=0.23; P <0.0001), respectively. The most common adverse reactions were fatigue, rash, nausea, vomiting, diarrhea, abdominal discomfort, cytopenias and abnormal liver function studies. Discontinuation due to adverse reactions occurred in 15% of patients receiving RUBRACA®.
It was concluded that RUBRACA® significantly improved Progression Free Survival in patients with platinum-sensitive ovarian cancer who had achieved a response to platinum-based chemotherapy, and could be considered a new standard of care for women with platinum-sensitive ovarian cancer, following a complete or partial response to second-line or later lines of platinum-based chemotherapy. The FDA also concurrently approved the complementary diagnostic test, FoundationFocusTM CDx BRCA LOH for tumor samples, to determine HRD status.
Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Coleman RL, Oza AM, Lorusso D, et al. The Lancet 2017;390:1949-1961
