
The FDA on January 15, 2021 granted accelerated approval to DARZALEX Faspro® in combination with Bortezomib, Cyclophosphamide and Dexamethasone for newly diagnosed light chain (AL) Amyloidosis. DARZALEX FASPRO® is a product of Janssen Biotech Inc.
Tag: Multiple Myeloma
Genomics Identify Patients with Smoldering Myeloma at Risk of Developing Multiple Myeloma
SUMMARY: Multiple Myeloma (MM) is a clonal disorder of plasma cells in the bone marrow. It evolves from a precursor stage called Monoclonal Gammopathy of Unknown Significance (MGUS) to MM. Smoldering Multiple Myeloma (SMM) is an intermediate stage in this process of disease evolution. The risk of MGUS transforming into MM is approximately 1% per year. Smoldering Multiple Myeloma or asymptomatic MM is a precursor to MM and is characterized by at least 10% plasma cells in the bone marrow or M-spike of at least 3 g/dl, or both, but these patients have no evidence of active symptomatic Myeloma with associated end-organ damage such as hypercalcemia, renal insufficiency, anemia or bone lesions. Even though only 10% of patients with SMM progress to MM annually, over 50% of the SMM patients with high risk features will progress to MM in the first 2 years.
The current recommendations for those with SMM are periodic monitoring and treatment intervention only when disease progresses to MM. SMM patients with high risk features include those with at least 10% plasma cells in the bone marrow, a Monoclonal component (IgG monoclonal spike of at least 3 g/dL, IgA M-spike of at least 2 g/dL or a urinary Bence Jones protein level of more than 1 g per 24 hours) or only one of the above two criteria plus at least 95% abnormal plasma cells in the bone marrow, with a reciprocal decrease in one or two uninvolved immunoglobulins of more than 25%, compared to normal values.
Identifying SMM patients who are at a high risk for progression to Multiple Myeloma can allow for early intervention to prevent end-organ damage and potentially achieve long-term remission. Current prognostic models rely solely on clinical markers and do not fully capture the risk of SMM progression. The authors in this study hypothesized that genetic alterations can predict the risk of progression from SMM to overt Multiple Myeloma (MM).
The researchers conducted a multicenter study on bone marrow samples from 214 patients at the time of diagnosis with SMM, using Next-Generation Sequencing (NGS) technologies. This study included an external validation cohort of 72 patients with SMM, whose tumor DNA has been previously sequenced. Whole-Exome Sequencing was performed on 166 tumor samples, and deep targeted sequencing on 48 tumor samples. This study excluded patients who presented at diagnosis with MM related findings such as hypercalcemia, renal impairment, anemia, or bone lytic lesions or who had any myeloma-defining event. Patients with light-chain and nonsecretory SMM were however included. The median patient age was 62 years. Patients were followed up for a median of 6.8 years to identify which of these patients developed myeloma, and the researchers then cross-linked the molecular and clinical data to explore whether certain genomic abnormalities increased the risk of progression to myeloma.
It was noted that most of the genetic alterations necessary for progression to MM were already present by the time of diagnosis of SMM and were all independent risk factors of progression, after accounting for clinical risk staging. They included alterations of the MAPK pathway (KRAS and NRAS Single Nucleotide Variants-SNVs), DNA repair pathway (deletion 17p, TP53, and ATM SNVs) and amplification or translocation of MYC gene.
Patients who harbored MYC aberrations (translocations or amplifications) had the shortest median Time to Progression (8.4 versus 51.6 months; P<0.001) followed by those with MAPK pathway mutations (14.4 versus 60 months; P<0.001) and DNA repair pathway alterations (15.6 versus 50.4 months; P=0.004). These findings were validated in the external cohort of 72 patients with SMM whose tumor DNA had been previously sequenced and the researchers found that patients with any of the high-risk genetic alterations also had a higher risk of progression to MM. APOBEC (“apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like”) associated mutations were enriched in patients who progressed to MM, and were associated with a shorter time to progression.
It was concluded that the genetic alterations with Smoldering Multiple Myeloma are essentially the same as full-fledged myeloma suggesting that by the time Smoldering Multiple Myeloma is diagnosed, most of the molecular abnormalities found in myeloma have already occurred. The authors added that genomic predictors of progression could identify patients at high risk of progression to Multiple Myeloma and thus improve on the precision of current clinical models. However, the role played by tumor microenvironment in the risk of disease progression, remains to be determined.
Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. Bustoros M, Sklavenitis-Pistofidis, Park J, et al. J Clin Oncol 2020;38:2380-2389.
FDA Approves XPOVIO® for Relapsed or Refractory Multiple Myeloma
SUMMARY: The FDA on December 18, 2020 approved XPOVIO® (Selinexor) in combination with VELCADE® (Bortezomib) and Dexamethasone for the treatment of adult patients with multiple myeloma who have received at least one prior therapy. Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 32,270 new cases will be diagnosed in 2020 and 12,830 patients are expected to die of the disease. Multiple Myeloma (MM) in 2020 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Multiple Myeloma is a disease of the elderly, with a median age at diagnosis of 69 years and characterized by intrinsic clonal heterogeneity. Almost all patients eventually will relapse, and patients with a high-risk cytogenetic profile or refractory disease have the worst outcomes. The median survival for patients with myeloma is over 10 years.
Exportin 1 (XPO1) is an important nuclear export protein overexpressed in multiple myeloma. High XPO1 levels facilitate increased nuclear export of tumor suppressor proteins such as P53, P73, IkB and FOXO3a, pRb, BRCA1, as well as growth regulators such as Glucocorticoid Receptor and oncoprotein mRNA. This enables cancer cells to escape tumor suppressor protein mediated cell cycle arrest and apoptosis. XPOVIO® is an oral selective XPO1inhibitor that reactivates the tumor suppressor proteins by preventing nuclear transport, inhibits oncoprotein translation and reactivates Glucocorticoid Receptor signaling in the presence of Dexamethasone. In a Phase Ib/II study, the combination of XPOVIO® along with VELCADE® (a Proteasome Inhibitor) and Dexamethasone induced high response rates with low rates of peripheral neuropathy.
The present FDA approval for XPOVIO® was based on findings from the BOSTON trial, which is a multicenter, open-label, randomized, controlled Phase III study, conducted to evaluate the clinical benefit of weekly XPOVIO®, VELCADE® (Bortezomib), and Dexamethasone, versus standard VELCADE® and Dexamethasone, in patients with previously treated multiple myeloma. In this study, 402 patients were randomly assigned 1:1 to receive either XPOVIO® 100 mg PO once weekly, VELCADE® 1.3 mg/m2 SC once weekly, and Dexamethasone 20 mg PO twice weekly, or VELCADE® 1.3 mg/m2 SC twice weekly for the first 24 weeks and once weekly thereafter, and Dexamethasone 20 mg four times per week for the first 24 weeks and twice weekly thereafter. The median patient age was 67 years and 32% of the patients had 2 prior lines of therapy, including prior REVLIMID® (Lenalidomide) in 38% and prior VELCADE® in 69%. Approximately 48% of the patients had high-risk cytogenetics which included del(17p), t(4;14), t(14;16) or amp(1q21). The Primary endpoint was Progression Free Survival (PFS), and Secondary endpoints included Objective Response Rate (ORR), Duration of Response (DoR), Overall Survival (OS) and Safety.
It was noted that the median PFS was 13.9 months in the XPOVIO® group and 9.5 months for the control group (HR=0.70; P=0.0075). This represented a 30% reduction in the risk of progression or death with the XPOVIO® triplet combination. This benefit was consistently noted across all subgroups including those with high-risk cytogenetics. The ORR was 76.4% in the XPOVIO® group versus 62.3% in the control group (P=0.0012), and the significantly higher ORR again was noted across subgroups. The median Duration of Response was 20.3 months versus 12.9 months in the XPOVIO® group and the control group, respectively. The most common adverse events in the XPOVIO® group included cytopenias, fatigue, nausea, diarrhea, asthenia, decreased appetite and weight loss.
It was concluded that weekly regimen of XPOVIO® given along with VELCADE® and Dexamethasone, is a novel, effective, and convenient treatment option, for patients with multiple myeloma, who have received one to three prior lines of therapy.
Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open-label, phase 3 trial. Grosicki S, Simonova M, Spicka I, et al. Lancet. 2020;396:1563-1573.
ASH 2020: Subcutaneous DARZALEX® Plus Pomalidomide and Dexamethasone in Relapsed or Refractory Multiple Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 32,270 new cases will be diagnosed in 2020 and 12,830 patients are expected to die of the disease. Multiple Myeloma (MM) in 2020 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Multiple Myeloma is a disease of the elderly, with a median age at diagnosis of 69 years and characterized by intrinsic clonal heterogeneity. Almost all patients eventually will relapse, and patients with a high-risk cytogenetic profile or refractory disease have the worst outcomes. The median survival for patients with Myeloma is over 10 years.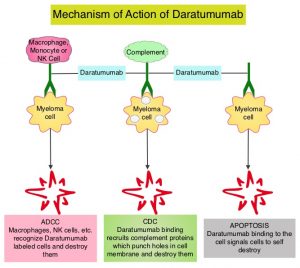
DARZALEX® is a human IgG1 antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Mediated Cytotoxicity and direct apoptosis. Additionally, DARZALEX® may have a role in immunomodulation by depleting CD38-positive regulator Immune suppressor cells, and thereby expanding T cells, in patients responding to therapy. DARZALEX® has activity as both a single agent and when combined with other standard regimens. POMALYST® (Pomalidomide) is a novel, oral, immunomodulatory drug which is far more potent than THALOMID® (Thalidomide) and REVLIMID®, and has been shown to be active in REVLIMID® and VELCADE® refractory patients. In the EQUULEUS Phase Ib study, intravenous DARZALEX® in combination with POMALYST® and Dexamethasone in relapsed or refractory Multiple Myeloma resulted in an Overall Response Rate (ORR) of 59% with Very Good Partial Response (VGPR) noted in 28% of patients, and Complete Response (CR) achieved in 6% of patients.
Recently published studies have concluded that the subcutaneous formulation of DARZALEX® resulted in non-inferior pharmacokinetics and efficacy compared to the current IV formulation, and also importantly offers the potential for a fixed-dose administration, shorter administration times and a lower rate of infusion-related reactions with improved safety profile.
APOLLO study is an open-label, randomized, multicenter, Phase III trial, conducted by the European Myeloma Network investigators, to evaluate SubCutaneous (SC) formulation of DARZALEX® in combination with POMALYST® (Pomalidomide) and Dexamethasone (D-Pd; N=151) versus POMALYST® (Pomalidomide) and Dexamethasone (Pd; N=153) alone in relapsed/refractory Multiple Myeloma patients who have received one or more prior lines of therapy including REVLIMID® (Lenalidomide) and a Proteasome Inhibitor. This study enrolled 304 patients with relapsed or refractory Multiple Myeloma, and prior treatment with anti-CD38 antibody or Pomalidomide was not permitted. Treatment for all patients consisted of POMALYST® 4 mg orally daily plus Dexamethasone 40 mg orally on days 1, 8, 15, and 22 (20 mg for patients aged 75 years or older), given every 28 days. Patients in the D-Pd group additionally received DARZALEX® 1800 mg SC co-formulated with recombinant human hyaluronidase PH20 (rHuPH20; ENHANZE® drug delivery technology, Halozyme, Inc.), given weekly for cycles 1 to 2, every 2 weeks for cycles 3 to 6, and every 4 weeks thereafter. The median age was 67 years, and 35% had high cytogenetic risk (presence of del17p, t[14;16], or t[4;14]). The median prior lines of therapy were 2, approximately 80% of patients were refractory to REVLIMID®, 48% of patients were refractory to a Proteosome Inhibitor, and 42% of patients were refractory to both agents. Treatment was continued until disease progression or unacceptable toxicity. The median duration of treatment was 11.5 months with D-Pd, compared with 6.6 months with Pd. The Primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Response Rate (ORR), Very Good Partial Response (VGPR), Complete Response (CR), MRD negativity rate, Overall Survival (OS), and Safety.
The study met its Primary endpoint of improved PFS in the primary analysis. The median PFS for the D-Pd group was 12.4 months versus 6.9 months for Pd group (HR=0.63; P=0.0018). This represented a 37% reduction in the risk of progression or death in patients treated with D-Pd. Among patients who were refractory to REVLIMID®, median PFS was 9.9 months in the D-Pd group versus 6.5 months in the Pd group. This benefit was seen across all subgroups of patients, regardless of age, stage, prior line of therapy, REVLIMID® refractoriness and cytogenetic risk. D-Pd regimen was also superior to Pd regimen in terms of other endpoints, including ORR (69% versus 46%), VGPR or better (51% versus 20%), CR (25% versus 4%), and MRD negativity (9% versus 2%). Survival data are immature and follow up is ongoing. Infusion-related events were rare, and seen in 6% of patients treated with D-Pd, and local injection-site reactions which were all Grade 1 were seen in 2% of patients in the D-Pd group. Treatment discontinuation due to treatment-related adverse events, were similar for the D-Pd and Pd groups (2% versus 3%).
It was concluded that Subcutaneous DARZALEX® given along with POMALYST® and Dexamethasone significantly reduced the risk of progression or death by 37% in patients with relapsed/refractory Multiple Myeloma, compared to POMALYST® and Dexamethasone alone. The infusion-related reaction rate was very low and median duration of injection administration was short at 5 minutes. Subcutaneous DARZALEX® thus has a high likelihood of changing clinical practice, increasing convenience for patients and decreasing treatment burden.
Apollo: Phase 3 Randomized Study of Subcutaneous Daratumumab Plus Pomalidomide and Dexamethasone (D-Pd) Versus Pomalidomide and Dexamethasone (Pd) Alone in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM). Dimopoulos MA, Terpos E, Boccadoro M, et al. Presented at the 62nd ASH Annual Meeting and Exposition, 2020. Abstract 412.
FDA Approves DARZALEX® plus KYPROLIS® and Dexamethasone for Multiple Myeloma
SUMMARY: The FDA on August 20, 2020, approved KYPROLIS® (Carfilzomib) and DARZALEX® (Daratumumab), in combination with Dexamethasone, for adult patients with Relapsed or Refractory multiple myeloma, who have received one to three lines of therapy. Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 32, 270 new cases will be diagnosed in 2020 and 12,830 patients are expected to die of the disease. Multiple Myeloma (MM) in 2020 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Multiple Myeloma is a disease of the elderly, with a median age at diagnosis of 69 years and characterized by intrinsic clonal heterogeneity. Almost all patients eventually will relapse, and patients with a high-risk cytogenetic profile or refractory disease have the worst outcomes. The median survival for patients with Myeloma is over 10 years.
REVLIMID® (Lenalidomide) in combination with VELCADE® (Bortezomib) and Dexamethasone is the preferred regimen according to the NCCN guidelines, for both transplant and non-transplant candidates with newly diagnosed Multiple Myeloma, and when given continuously or with maintenance therapy, has improved survival outcomes. Nonetheless, a significant number of patients progress while on these agents or discontinue therapy due to toxicities. There is therefore a need for effective and tolerable regimens for patients who are exposed or refractory to REVLIMID® or VELCADE®.
KYPROLIS® (Carfilzomib) is a second generation selective, epoxyketone Proteasome Inhibitor and unlike VELCADE®, proteasome inhibition with KYPROLIS® is irreversible. DARZALEX® (Daratumumab) is a human IgG1 antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Dependent Cytotoxicity (CDC) and direct Apoptosis. Additionally, DARZALEX® may play a role in immunomodulation, by depleting CD38-positive regulator immune suppressor cells, and thereby expanding T cells, in patients responding to therapy. Both KYPROLIS® and DARZALEX® are approved as single agents, as well as in combination with other drugs, for the treatment of patients with Relapsed/Refractory Multiple Myeloma. In a Phase I study, KYPROLIS® in combination with Dexamethasone and DARZALEX® demonstrated safety and efficacy in patients Relapsed/Refractory Multiple Myeloma.
The efficacy of KYPROLIS® and DARZALEX® along with Dexamethasone was evaluated in two clinical trials, CANDOR and EQUULEUS. CANDOR is a multicenter, open-label, Phase III trial, which included Relapsed/Refractory Multiple Myeloma patients with measurable disease who had received 1-3 prior lines of therapy, with Partial Response or better to one or more lines of therapy. A total of 466 patients were randomly assigned 2:1 to receive triplet of KYPROLIS®, Dexamethasone, and DARZALEX® (KdD)- N=312 or KYPROLIS® and Dexamethasone (Kd) alone- N=154. All patients received KYPROLIS® as a 30 minute IV infusion on days 1, 2, 8, 9, 15, and 16 of each 28-day cycle (20 mg/m2 on days 1 and 2 during cycle 1 and 56 mg/m2 thereafter). DARZALEX® 8 mg/kg was administered IV on days 1 and 2 of cycle 1 and at 16 mg/kg once weekly for the remaining doses of the first 2 cycles, then every 2 weeks for 4 cycles (cycles 3-6), and every 4 weeks thereafter. All patients received Dexamethasone 40 mg oral or IV weekly (20 mg for patients over 75 years of age). The median age was 64 years, 42% and 90% received prior REVLIMID® and VELCADE® (Bortezomib) containing regimens respectively, and a third of patients were refractory to REVLIMID®. The Primary endpoint was Progression Free Survival (PFS) and Secondary endpoints included Overall Response Rate (ORR), Minimal Residual Disease (MRD)-negative status, Complete Response (CR) rate at 12 months, Overall Survival (OS), Duration of Response, and Safety.
After a median follow up of 17 months, the study met its Primary endpoint and the median PFS was not reached for the KdD arm and was 15.8 months for the Kd arm (HR=0.63; P=0.0027). This represented a 37% reduction in the risk of progression or death in the KdD group. The PFS benefit of KdD was maintained across prespecified subgroups, particularly among REVLIMID®-exposed and REVLIMID®-refractory patients. The ORR was 84.3% in the KdD group versus 74.7% in the Kd group (P=0.004), with a CR rate or better of 28.5% versus 10.4% respectively. The median time to first response was one month in both treatment groups. Patients treated with KdD achieved deeper responses which was nearly 10 times higher, with a MRD-negative Complete Response rate at 12 months of 12.5% for KdD versus 1.3% for Kd (P<0.0001). The median treatment duration was longer in the KdD group compared to the Kd group (70.1 versus 40.3 wks). The median OS was not reached in either groups, at a median follow up time of 17 months. Toxicities were generally manageable and the incidence of Adverse Events leading to treatment discontinuation was similar in both treatment groups.
EQUULEUS is an open label, multicohort trial which evaluated the combination of KYPROLIS® administered weekly on days 1, 8, and 15 of each 28-day cycle (20 mg/m2 IV on cycle 1, day 1, and if tolerated, increased to 70 mg/m2 on Cycle 1 Day 8 and thereafter) along with DARZALEX® IV and Dexamethasone (KdD). Efficacy was based on Overall Response Rate (ORR). Of the 85 patients with Relapsed or Refractory multiple myeloma who had received 1 to 3 prior lines of therapy enrolled in the KdD cohort, the ORR was 81%, with response duration of 27.5 months.
It was concluded that a combination of KYPROLIS® along with Dexamethasone and DARZALEX® resulted in a significant PFS benefit over KYPROLIS® and Dexamethasone alone, with deeper responses, and the PFS benefit of KdD was maintained across prespecified, clinically important subgroups, particularly REVLIMID®-exposed and REVLIMID®-refractory patients. The authors added that KdD regimen should be considered as a novel, efficacious, and tolerable immunomodulatory-free treatment option for Relapsed/Refractory Multiple Myeloma patients.
Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Dimopoulos M, Quach H, Mateos M-V, et al. The Lancet 2020;396:186-197.
KYPROLIS® and DARZALEX®
The FDA on August 20, 2020, approved KYPROLIS® (Carfilzomib) and DARZALEX® (Daratumumab) in combination with Dexamethasone for adult patients with Relapsed or Refractory multiple myeloma who have received one to three lines of therapy. KYPROLIS® is a product of Onyx Pharmaceuticals, Inc. DARZALEX® is a product of Janssen Biotech, Inc.
BLENREP® (Belantamab mafodotin-blmf)
The FDA on August 5, 2020 approved BLENREP® for adult patients with Relapsed or Refractory Multiple Myeloma who have received at least 4 prior therapies, including an anti-CD38 monoclonal antibody, a Proteasome Inhibitor, and an Immunomodulatory agent. BLENREP® is a product of GlaxoSmithKline.
FDA Approves Subcutaneous DARZALEX® in Multiple Myeloma
SUMMARY: The FDA on May 1, 2020 approved DARZALEX® (Daratumumab) and Hyaluronidase-fihj (DARZALEX FASPRO®), for adult patients with newly diagnosed or Relapsed/Refractory multiple myeloma. This new product allows for subcutaneous dosing of DARZALEX®.
DARZALEX FASPRO® is now approved for these previously approved indications for IV DARZALEX®
1) In combination with VELCADE® (Bortezomib), Melphalan and Prednisone in newly diagnosed patients who are ineligible for Autologous Stem Cell Transplant (ASCT)
2) In combination with REVLIMID® (Lenalidomide) and Dexamethasone in newly diagnosed patients, who are ineligible for ASCT and in patients with Relapsed or Refractory multiple myeloma who have received at least one prior therapy
3) In combination with VELCADE® and Dexamethasone in patients who have received at least one prior therapy
4) As monotherapy, in patients who have received at least three prior lines of therapy including a Proteasome Inhibitor (PI) and an Immunomodulatory agent or who are double-refractory to a PI and an immunomodulatory agent.
DARZALEX® is a human IgG1 antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Dependent Cytotoxicity (CDC) and direct Apoptosis. Additionally, DARZALEX® may play a role in immunomodulation, by depleting CD38-positive regulator immune suppressor cells, and thereby expanding T cells, in patients responding to therapy.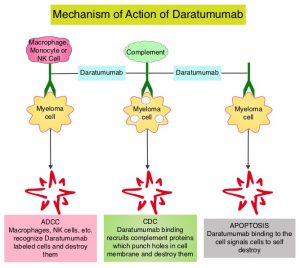
This FDA approval is based on COLUMBA Trial, which is a randomized, open-label, multicenter Phase III study, which included 522 patients with multiple myeloma, who had received at least three prior lines of therapy including a Proteasome Inhibitor (PI) and an immunomodulatory drug (IMiD), or whose disease was refractory to both a PI and an IMiD. Patients were randomly assigned to receive a fixed dose of subcutaneously (SC) administered formulation of DARZALEX® 1800 mg weekly for cycles 1-2, every two weeks for cycles 3-6 and every four weeks for cycle 7 and thereafter (N=263), with the subcutaneous preparation given over 3-5 minutes at alternating left and right abdominal sites. In the intravenous group, patients received DARZALEX® 16 mg/kg IV weekly for cycles 1-2, every two weeks for cycles 3-6 and every four weeks for cycle 7 and thereafter (N=259). Each cycle was 28 days. Treatment in both patient groups was continued until disease progression or unacceptable toxicity. The median age was 67 years and the median number of prior therapies was four in each treatment group. Patient characteristics were similar between the two arms except that more patients in the subcutaneous arm had high-risk cytogenetics (26%) compared with the intravenous group (17%). The median duration of treatment was approximately 5 months, with a median of 6 completed cycles of treatment. The median duration of infusion was consistently 5 minutes at each visit in the subcutaneous group. However, in the IV arm, the first infusion lasted 7 hours, the second infusion was 4.3 hours, and subsequent infusions lasted a median of 3.4 hours. The study co-Primary endpoints were Overall Response Rate (ORR) and pharmacokinetic endpoint of the maximum C-trough on cycle 3, day 1 pre-dose.
At a median follow up of 7.5 months, the ORR was 41% for the subcutaneous administered formulation of DARZALEX® compared to 37% for IV DARZALEX® (P<0.0001). The ORR was similar across all clinically relevant subgroups, including body weight. The ratio of geometric means of C-trough for the SC administered formulation of DARZALEX® over IV DARZALEX® was 108%. The Progression Free Survival was comparable between the SC administered formulation of DARZALEX and the current IV formulation of DARZALEX (HR=0.99; P<0.9258). A lower rate of infusion-related reactions was observed in the group that received the SC DARZALEX® compared to IV DARZALEX® (13% vs. 35%, respectively).
It was concluded that the subcutaneous formulation of DARZALEX® resulted in non-inferior pharmacokinetics and efficacy compared to the current IV formulation, and also importantly offers the potential for a fixed-dose administration, shorter administration times and a lower rate of infusion-related reactions with improved safety profile, in patients with Relapsed or Refractory multiple myeloma.
Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): a multicentre, open-label, non-inferiority, randomised, phase 3 trial. Mateos M-V, Nahi H, Legiec W, et al. The Lancet Haematology. Published: March 23, 2020. DOI: https://doi.org/10.1016/S2352-3026(20)30070-3.
DARZALEX FASPRO® (Daratumumab and Hyaluronidase-fihj)
The FDA on May 1, 2020 approved DARZALEX FASPRO® for adult patients with newly diagnosed or Relapsed/Refractory Multiple Myeloma. This new product allows for subcutaneous dosing of Daratumumab. DARZALEX FASPRO® is a product of Janssen Biotech, Inc.