SUMMARY: Chemotherapy remains one of the mainstays of cancer treatment. However, chemotherapy-induced damage of Hematopoietic Stem and Progenitor Cells (HSPC) causes multi-lineage myelosuppression. Currently, available therapies such as Granulocyte-Colony Stimulating Factors (G-CSF) and Erythropoiesis-Stimulating Agents (ESAs) prevent the myelosuppressive effects of chemotherapy in only one lineage. Therapeutic agents that lead to protection of multiple lineages simultaneously would be clinically meaningful.
Trilaciclib is a highly potent, selective, and reversible, intravenous, Cyclin-Dependent Kinase 4 and 6 (CDK 4/6) inhibitor, that transiently maintains G1 cell cycle arrest of Hematopoietic Stem and Progenitor Cells (HSPC), and protects them from damage by cytotoxic chemotherapy. Chemotherapy-induced damage of Hematopoietic Stem and Progenitor Cells (HSPC) causes multi-lineage myelosuppression. Trilaciclib proactively preserves HSPC and immune system function during chemotherapy (myelopreservation). Preclinical studies have demonstrated that Trilaciclib transiently maintains HSPC in G1 arrest and protects them from chemotherapy damage, leading to faster hematopoietic recovery. Additionally, Trilaciclib enhances immune response, and preserves immune system function.
Small Cell Lung Cancer (SCLC) was chosen as the testing platform, to explore the potential myelopreservation benefit of Trilaciclib for the following reasons: 1) Cytotoxic chemotherapy for SCLC is notable for its myelotoxicity. 2) SCLC tumor cells replicate independent of CDK4/6, through the obligate loss of Retinoblastoma (RB1), allowing assessment of Trilaciclib’s effects on the host, without any potential direct effects on the tumor. 3) SCLC is a chemosensitive tumor, and provides an optimal setting to demonstrate that Trilaciclib does not antagonize chemotherapy efficacy.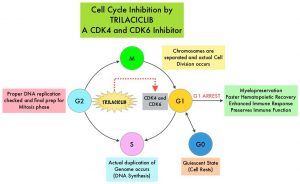
The authors in this publication, pooled data from three randomized, double-blind, placebo-controlled Phase II trials, in patients with Extensive-Stage Small Cell Lung Cancer (ES-SCLC), to understand the effects of Trilaciclib on specific myelosuppression endpoints, with greater statistical precision. Individual results from these three randomized trials have previously been reported. In this pooled analysis, 123 patients received Trilaciclib along with chemotherapy (N=123), and 119 patients received Placebo along with chemotherapy (N=119). The median age in both treatment groups was 64 years. The objectives of this pooled data analysis was to summarize the utilization of G-CSFs, ESAs and RBC transfusions, and hospitalizations due to Chemotherapy Induced Myelosuppression or sepsis, as well as explore the relationship between supportive care interventions and the myelopreservation benefits of Trilaciclib.
In the first study (NCT02499770), patients with newly diagnosed ES-SCLC received Trilaciclib 240 mg/m2 or Placebo IV, given daily on days 1 to 3, prior to chemotherapy, of each 21-day chemotherapy cycle with Etoposide and Carboplatin. In the second trial (NCT03041311), patients with newly diagnosed ES-SCLC received Trilaciclib 240 mg/m2 or Placebo IV, given daily on days 1 to 3, prior to chemotherapy, of each 21-day chemotherapy cycle with Etoposide, Carboplatin and Atezolizumab (TECENTRIQ®), followed by single-agent Atezolizumab alone, every 21 days. In the third study (NCT02514447), patients with previously treated ES-SCLC in the second or third line setting, received Trilaciclib 240 mg/m2 or Placebo IV daily, prior to Topotecan 1.5 mg/m2 IV given daily on days 1 to 5 of each 21-day cycle. The Primary outcome measures included percentage of patients with Severe (Grade 4) Neutropenia as well as duration of Severe Neutropenia. Supportive intervention endpoints included percentage of patients with RBC transfusions on or after week 5, and number of RBC transfusion events on or after week 5, as well as percentage of patients receiving ESAs. This study also explored the percentage of patients with hospitalizations due to Chemotherapy Induced Myelosuppression (neutropenia, anemia, thrombocytopenia) or sepsis, as well as incidence of hospitalizations due to Chemotherapy Induced Myelosuppression or sepsis, per 100 cycles.
It was noted that fewer patients receiving Trilaciclib had Severe Neutropenia (11.4% versus 52.9%) or Grade 3/4 anemia (20.3% versus 31.9%), compared to Placebo, respectively, and the use of supportive care interventions such as G-CSF and ESAs was significantly reduced. Hospitalizations due to Chemotherapy Induced Myelosuppression or sepsis occurred in significantly fewer patients and significantly less often among patients receiving Trilaciclib prior to chemotherapy, compared to those who received Placebo. Trilaciclib reduced the percentage of patients with Severe Neutropenia and duration of Severe Neutropenia, regardless of G-CSF administration. The proportion of patients receiving RBC transfusions was consistently lower with each cycle, among patients receiving Trilaciclib, whereas RBC transfusions in the Placebo group almost doubled over time.
It was concluded that Trilaciclib prior to chemotherapy significantly and meaningfully reduced Chemotherapy Induced Myelosuppression and the need for supportive care interventions, for the management of Severe Neutropenia and Grade 3/4 anemia, in patients with ES-SCLC. Chemotherapy-induced Severe Neutropenia was reduced with Trilaciclib, irrespective of G-CSF administration.
Trilaciclib Reduces the Need for Growth Factors and Red Blood Cell Transfusions to Manage Chemotherapy-Induced Myelosuppression. Ferrarotto R, Anderson I, Medgyasszay B, et al. Presented at: IASLC 2020 North America Conference on Lung Cancer; October 16-17, 2020.