
SUMMARY: The FDA on November 30, 2017, granted marketing approval to FoundationOne CDx (F1CDx), a Next Generation Sequencing (NGS) based, in vitro diagnostic (IVD) assay, to detect genetic mutations in 324 genes and two genomic signatures, in any solid tumor type. The test can also identify which patients with Non Small Cell Lung Cancer (NSCLC), Melanoma, Breast cancer, ColoRectal cancer, or Ovarian cancer may benefit from 15 different FDA-approved targeted treatment options.
The basic premise of cancer genomics is that cancer is caused by somatically acquired mutations, and is therefore a disease of the genome. Tumor genomic profiling enables the identification of specific genomic alterations and thereby can provide personalized treatment options with targeted therapies that are specific for those molecular targets. A genomic test can be performed on a tumor specimen or on cell-free DNA in plasma (“liquid biopsy”) or an ImmunoHistoChemistry (IHC) test can be performed on tumor tissue for protein expression that demonstrates a genomic variant known to be a drug target, or to predict sensitivity to a chemotherapeutic drug.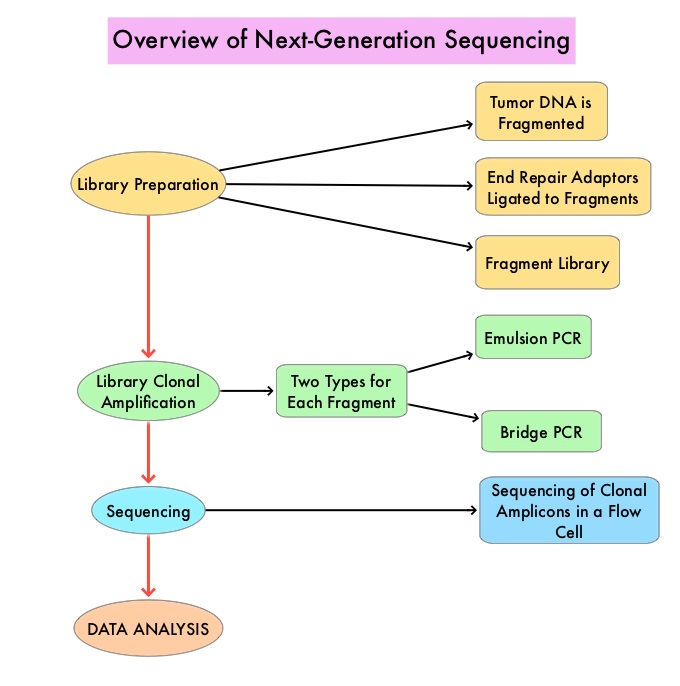
Next-Generation Sequencing (NGS) platforms or second-generation sequencing, unlike the first-generation sequencing, known as Sanger sequencing, perform massively parallel sequencing, which allows sequencing of millions of fragments of DNA from a single sample. With this high-throughput sequencing, the entire genome can be sequenced in less than 24 hours. This is in contrast to Sanger sequencing technology which has required over a decade to decipher the human genome. There are a number of different NGS platforms using different sequencing technologies and NGS can be used to sequence and systematically study the cancer genomes in their entirety or specific areas of interest in the genome or small numbers of individual genes. Recently reported genomic profiling studies, performed in patients with advanced cancer suggest that actionable mutations are found in 20-40% of patients’ tumors.
The application for F1CDx , was reviewed by the FDA using a coordinated, cross-agency approach and clinical performance of the test was established by comparing F1CDx to previously FDA-approved companion diagnostic tests, that are currently used to determine patient eligibility for certain treatments. It was noted that F1CDx assay’s ability to detect select mutation types (substitutions and short insertions and deletions) representative of the entire 324 gene panel was accurate approximately 94.6% of the time. This 324 gene panel included EGFR, KRAS, BRAF, BRCA1/2, ALK, and several other genes with emerging therapies, such as NTRK1/2/3. This assay can additionally detect MicroSatellite Instability (MSI) and Tumor Mutational Burden, which can predict response to immunotherapy.
The FDA noted that this is the first device with the FDA’s “Breakthrough Device” designation to complete the PreMarket Approval (PMA) process, and it is the second IVD authorized under the FDA and Centers for Medicare & Medicaid Services’ (CMS) Parallel Review program. Under this program, the CMS issued a proposed national coverage determination of the F1CDx for Medicare beneficiaries with recurrent, metastatic, or advanced Stage IV cancer, who have not been previously tested using NGS technology, and who continue to remain candidates for further therapy. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm587387.htm
