SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers.
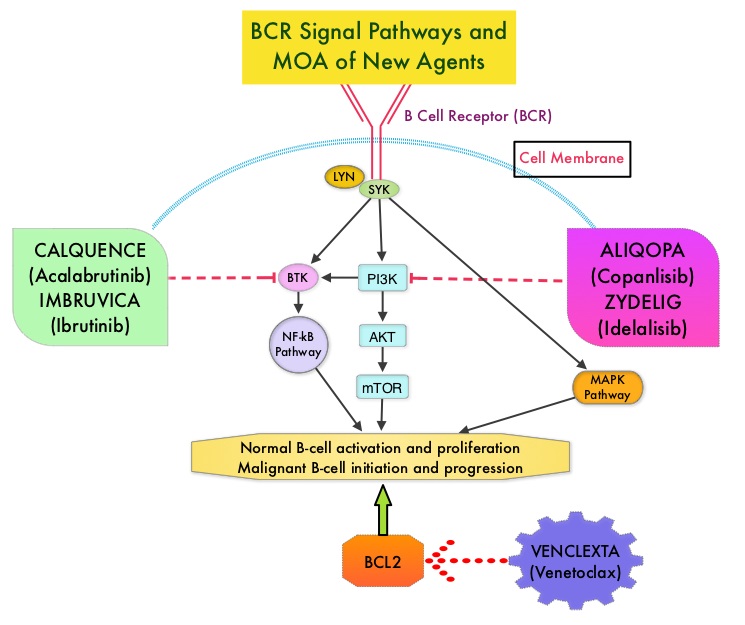
KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor (immune checkpoint protein) and blocks its interaction with ligands PD-L1 and PD-L2. This leads to the undoing of the PD-1 pathway-mediated inhibition of the immune response and the tumor-specific effector T cells are unleashed. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced Non Small Cell Lung Cancer (NSCLC) have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.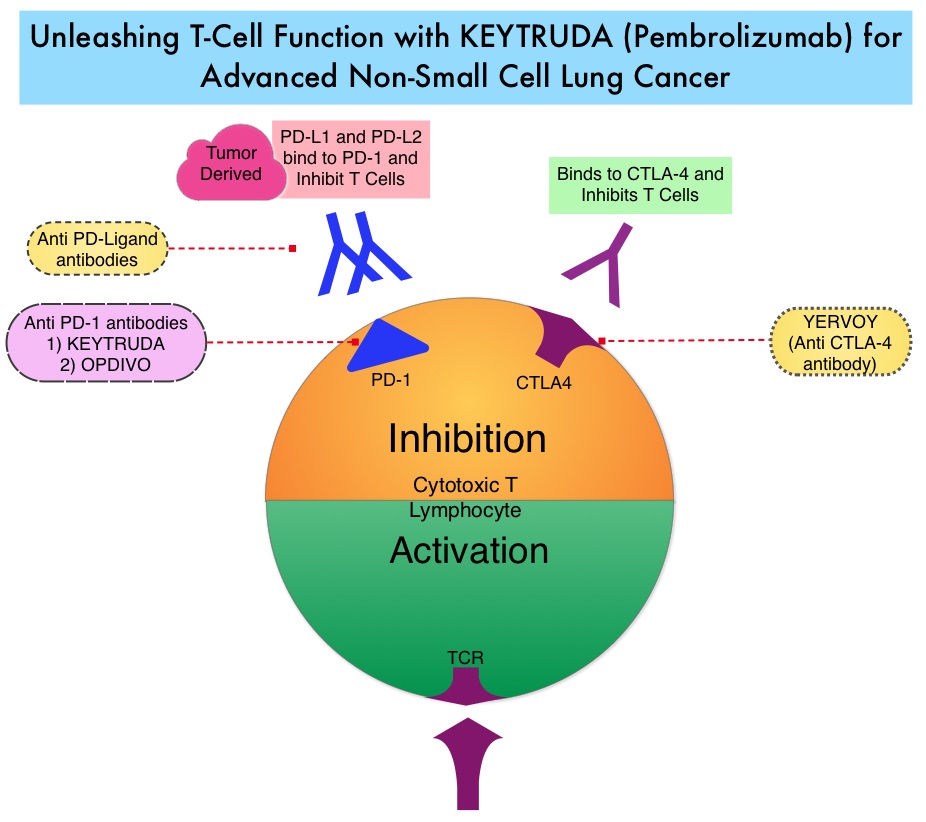
KEYNOTE-024 is an open-label, randomized phase III trial in which KEYTRUDA® administered at a fixed dose was compared with investigator’s choice of cytotoxic chemotherapy, as first line therapy, for patients with advanced NSCLC, with tumor PD-L1 expression of 50% or greater. Three hundred and five (N=305) treatment naïve patients with advanced NSCLC and PD-L1 expression on at least 50% of tumor cells, were randomly assigned in a 1:1 ratio to receive either KEYTRUDA® (N=154) or chemotherapy (N=151). Enrolled patients had no sensitizing EGFR mutations or ALK translocations. Treatment consisted of KEYTRUDA® administered at a fixed dose of 200 mg IV every 3 weeks for 35 cycles or the investigator’s choice of platinum-based chemotherapy for 4-6 cycles. Pemetrexed (ALIMTA®) based therapy was permitted only for patients who had non-squamous tumors and these patients could receive ALIMTA® maintenance therapy after the completion of combination chemotherapy. Patients in the chemotherapy group who experienced disease progression were allowed to cross over to the KEYTRUDA® group. The primary end point was Progression Free Survival (PFS) and secondary end points included Overall Survival (OS), Objective Response Rate (ORR) and safety.
It was previously reported that at a median follow up of 11.2 months, the median PFS was 10.3 months in the KEYTRUDA® group versus 6 months in the chemotherapy group (HR=0.50; P<0.001). However, median OS had not been reached in the KEYTRUDA® group at the time of that analysis. This publication is an updated analysis of the KEYNOTE-024 study, after a median follow-up of 25.2 months. Eighty two patients (N=82) assigned to chemotherapy, met criteria to cross over to the KEYTRUDA® group, upon progression. The median OS was 30 months in the KEYTRUDA® group and 14.2 months in the chemotherapy group (HR=0.63). Further, more patients in the KEYTRUDA® group achieved 12-month OS (70.3% vs. 54.8%) and an ORR response (45.5% vs. 29.8%), compared to the chemotherapy group. The ORR among patients who crossed over to KEYTRUDA®, was 20.7%. The median Duration of Response has not yet been reached for patients assigned to KEYTRUDA® and also for those who crossed over to KEYTRUDA®. For those assigned chemotherapy, the median Duration of Response was 7.1 months. Patients in the KEYTRUDA® group had lower rates of grade 3 to 5 adverse events compared to those in the chemotherapy group (31.2% vs 53.3%), as well as a lower rate of any-grade adverse events (76.6% vs 90%).
It was concluded that first-line treatment with KEYTRUDA® resulted in a significantly longer median OS with lower rates of Adverse Events, when compared to chemotherapy, among patients with metastatic NSCLC and high PD-L1 expression. Brahmer JR, Rodriguez-Abreu D, Robinson A, et al. Updated analysis of KEYNOTE-024: pembrolizumab vs platinum-based chemotherapy for advanced NSCLC with PD-L1 TPS>50%. Presented at: International Association for the Study of Lung Cancer 18th World Conference on Lung Cancer; Yokohama, Japan: October 15-18, 2017. Abstract OA 17.06.
 This new approval for BOSULIF® was based on the efficacy and safety data from BFORE trial, which is an ongoing randomized, open-label, multicenter, phase III study in which in 487 patients with Ph+ newly diagnosed Chronic Phase CML were randomized to receive either BOSULIF® 400 mg once daily (N=246) or GLEEVEC® (Imatinib) 400 mg once daily (N=241).
This new approval for BOSULIF® was based on the efficacy and safety data from BFORE trial, which is an ongoing randomized, open-label, multicenter, phase III study in which in 487 patients with Ph+ newly diagnosed Chronic Phase CML were randomized to receive either BOSULIF® 400 mg once daily (N=246) or GLEEVEC® (Imatinib) 400 mg once daily (N=241).