
SUMMARY: The U.S. FDA on March 6, 2015 approved ZARXIO® (Filgrastim-sndz), the first biosimilar product approved in the United States. A biosimilar product is a biological product that is approved based on its high similarity to an already approved biological product (also known as reference product). Biological products are made from living organisms including humans, animals and microorganisms such as bacteria or yeast and are manufactured through biotechnology, derived from natural sources or produced synthetically. Biological products have larger molecules with a complex structure than conventional drugs (also known as small molecule drugs). Unlike biological products, conventional drugs are made of pure chemical substances and their structures can be identified. A generic drug is a copy of brand name drug and has the same active ingredient and is the same as brand name drug in dosage form, safety and strength, route of administration, quality, performance characteristics and intended use. Therefore, brand name and the generic drugs are bioequivalent.
The Affordable Care Act in 2010 created an abbreviated licensure pathway for biological products that are demonstrated to be “biosimilar” to, or “interchangeable” with an FDA-licensed (FDA approved) biological product (reference product). The biosimilar must show that it has no clinically meaningful differences in terms of safety and effectiveness from the reference product. A biosimilar product can only be approved by the FDA if it has the same mechanism of action, route of administration, dosage form and strength as the reference product, and only for the indications and conditions of use that have been approved for the reference product. Biosimilars are not as easy to manufacture as generics (copies of brand name drugs) because of the complexity of the structure of the biologic product and the process used to make a biologic product. The facilities where biosimilars are manufactured must also meet the FDA’s standards.
The FDA’s approval of ZARXIO® was based on review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data and other clinical safety and effectiveness data, that demonstrated ZARXIO® was biosimilar to NEUPOGEN®. ZARXIO® was approved as a biosimilar and not as an interchangeable product (Can only be substituted for the reference product after approval by the prescribing Health Care Provider). ZARXIO® is approved for the same indications as NEUPOGEN® and these indications include
• Patients with cancer receiving myelosuppressive chemotherapy
• Patients with Acute Myeloid Leukemia receiving induction or consolidation chemotherapy
• Patients with cancer undergoing Bone Marrow Transplantation
• Patients undergoing Autologous peripheral blood progenitor cell collection and therapy
• Patients with severe Chronic Neutropenia.
The most common expected side effects of ZARXIO® are bone and muscle aches, redness, swelling or itching at injection site. Less common, serious side effects include spleen rupture and serious allergic reactions. Unlike ZARXIO® which was approved via an abbreviated licensure pathway for biosimilars, GRANIX® (tbo-Filgrastim) was approved via the full Biologic License Application pathway, which presently limits GRANIX® use only for reducing the duration of severe neutropenia in patients non-myeloid malignancies, receiving myelosuppressive chemotherapy. The present Medicare reimbursement rules will be more favorable to ZARXIO® compared to GRANIX®, based on their approval process. FDA approves first biosimilar product ZARXIO®. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm
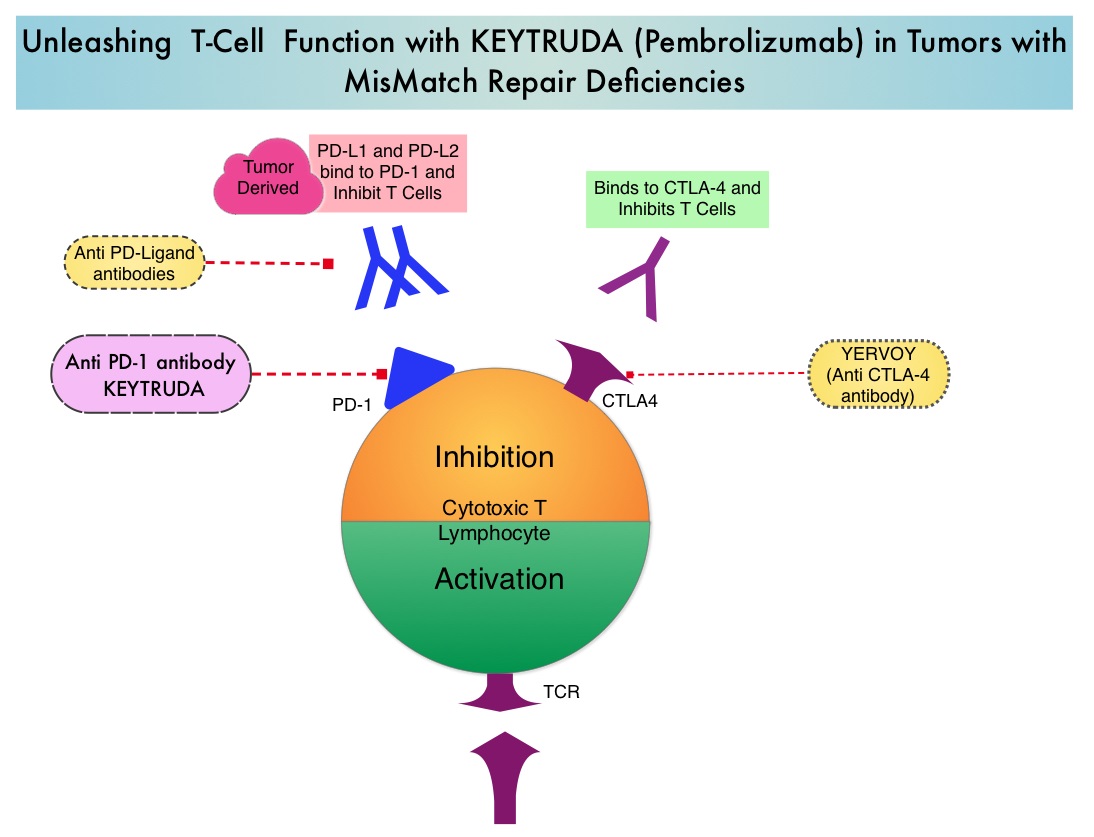 Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4), also known as CD152, PD-1(Programmed cell Death-1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic respons
Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4), also known as CD152, PD-1(Programmed cell Death-1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic respons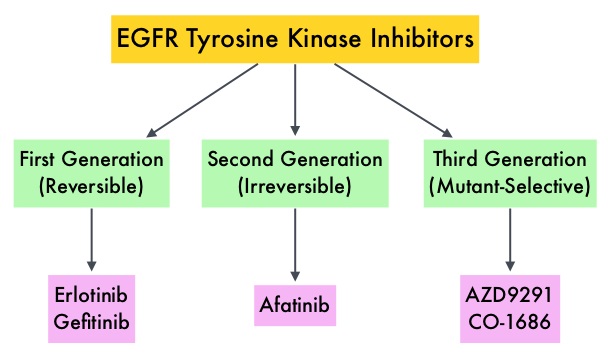
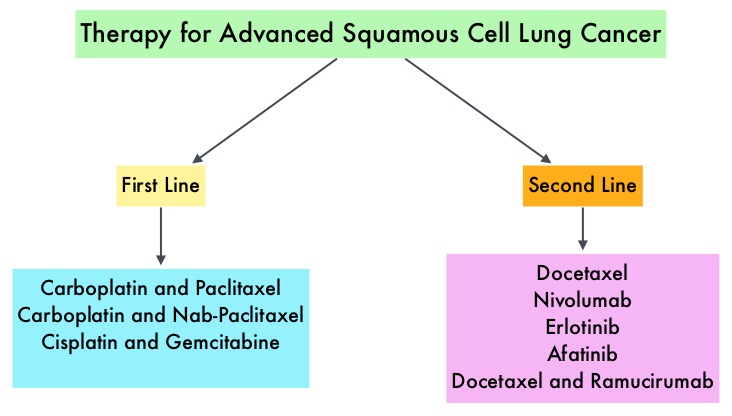 Non Small Cell Lung Cancer patients with squamous cell histology have been a traditionally hard- to-treat patient group, with less than 5% of patients with advanced SCC, surviving for five years or longer. Some of the advanced NSCLC tumors are dependent on the Epidermal Growth Factor Receptor (EGFR) for cell proliferation and survival, regardless of EGFR mutation status. TARCEVA® (Erlotinib) is a reversible EGFR Tyrosine Kinase Inhibitor and is presently approved by the FDA for the treatment of locally advanced or metastatic NSCLC, after failure of at least one prior chemotherapy regimen. GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. This kinase inhibitor is indicated for the first line treatment of patients with metastatic NSCLC, whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations.
Non Small Cell Lung Cancer patients with squamous cell histology have been a traditionally hard- to-treat patient group, with less than 5% of patients with advanced SCC, surviving for five years or longer. Some of the advanced NSCLC tumors are dependent on the Epidermal Growth Factor Receptor (EGFR) for cell proliferation and survival, regardless of EGFR mutation status. TARCEVA® (Erlotinib) is a reversible EGFR Tyrosine Kinase Inhibitor and is presently approved by the FDA for the treatment of locally advanced or metastatic NSCLC, after failure of at least one prior chemotherapy regimen. GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. This kinase inhibitor is indicated for the first line treatment of patients with metastatic NSCLC, whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations. Gleevec® (Imatinib) inhibits the BCR-ABL tyrosine kinase and is the standard first line treatment, of Ph chromosome positive (Ph+) leukemias. Lack of response due to resistance to GLEEVEC® and in some instances drug intolerance, has led to the development of newer agents including Second and Third generation Tyrosine Kinase Inhibitors (TKIs). Resistance to Gleevec® and other TKIs sharing the same therapeutic target (BCR-ABL kinase), has been attributed to point mutations in the ABL kinase domain, amplification of the BCR-ABL gene as well as other BCR- ABL independent mechanisms such as upregulation of SRC kinases. Mutation analysis at the time of TKI failure, utilizing high sensitivity sequencing techniques such as Next Generation Sequencing, can give clinically relevant information related to low level mutations and compound mutations and this information in turn, can dictate choice of second line therapy.
Gleevec® (Imatinib) inhibits the BCR-ABL tyrosine kinase and is the standard first line treatment, of Ph chromosome positive (Ph+) leukemias. Lack of response due to resistance to GLEEVEC® and in some instances drug intolerance, has led to the development of newer agents including Second and Third generation Tyrosine Kinase Inhibitors (TKIs). Resistance to Gleevec® and other TKIs sharing the same therapeutic target (BCR-ABL kinase), has been attributed to point mutations in the ABL kinase domain, amplification of the BCR-ABL gene as well as other BCR- ABL independent mechanisms such as upregulation of SRC kinases. Mutation analysis at the time of TKI failure, utilizing high sensitivity sequencing techniques such as Next Generation Sequencing, can give clinically relevant information related to low level mutations and compound mutations and this information in turn, can dictate choice of second line therapy.  The Second generation TKIs, TASIGNA® (Nilotinib) and SPRYCEL® (Dasatinib) although initially approved for second line treatment of CML after GLEEVEC® resistance or intolerance, are now FDA approved for the treatment of newly diagnosed Chronic Phase CML. This approval was based on the rapid and superior Major Molecular Responses (MMR) noted, when compared to GLEEVEC®. Now, that the Second generation TKIs are being used as first line therapy, the choice of second line therapy after failure with Second generation TKIs has become more nebulous. It is clear however that, patients with primary cytogenetic resistance to ï¬rst and second line therapy do not beneï¬t from sequential therapy with Second generation TKIs and BCR-ABL mutation analysis should be performed in all patients who develop TKI resistant disease. Before switching from a Second to a Third generation TKI such as Ponatinib, the following considerations should be taken into account
The Second generation TKIs, TASIGNA® (Nilotinib) and SPRYCEL® (Dasatinib) although initially approved for second line treatment of CML after GLEEVEC® resistance or intolerance, are now FDA approved for the treatment of newly diagnosed Chronic Phase CML. This approval was based on the rapid and superior Major Molecular Responses (MMR) noted, when compared to GLEEVEC®. Now, that the Second generation TKIs are being used as first line therapy, the choice of second line therapy after failure with Second generation TKIs has become more nebulous. It is clear however that, patients with primary cytogenetic resistance to ï¬rst and second line therapy do not beneï¬t from sequential therapy with Second generation TKIs and BCR-ABL mutation analysis should be performed in all patients who develop TKI resistant disease. Before switching from a Second to a Third generation TKI such as Ponatinib, the following considerations should be taken into account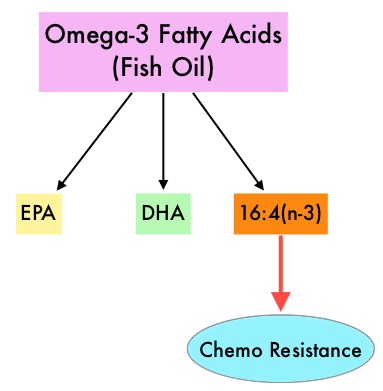 Fish oil has relevant levels of fatty acid 16:4(n-3) and preclinical models have shown that the fish oil neutralized the antitumor activity of chemotherapy, thus conferring drug resistance. With this preclinical information and given that cancer patients frequently use fish oil supplements, the authors evaluated the effect of fish oil intake in healthy volunteers, on the plasma levels of fatty acid 16:4(n-3), which has been shown to induce resistance to chemotherapeutic agents. The researchers first conducted a survey to determine what percentage of cancer patients undergoing treatment at a University Medical Center in the Netherlands were taking fish oil supplements. They also analyzed fatty acid 16:4(n-3) content, in 3 brands of fish oil supplements and 4 often consumed species of fish. The authors then randomly selected 30 healthy volunteers for the fish oil study and 20 healthy volunteers for the fish consumption study and the plasma levels of fatty acid 16:4(n-3) was measured after they consumed fish oil or fish, for a period of 2 weeks. They noted that 11% of the cancer patients in their study reported using omega-3 supplements. All fish oils tested contained amounts of fatty acid 16:4(n-3) ranging from 0.2 to 5.7 μM and this was adequate to induce chemoresistance to a variety of chemotherapeutic agents. They noted that there was a significant rise in the plasma 16:4(n-3) fatty acid levels in the healthy volunteers after they consumed fish oil supplements and fish, with high levels of fatty acid 16:4(n-3). Herring and Mackerel fish contained high levels of fatty acid 16:4(n-3), in contrast to Salmon and Tuna. The authors concluded that based on this preclinical data it is best to avoid fish oils and fish such as Herring and Mackerel in the 48 hours surrounding chemotherapy, as the high plasma 16:4(n-3) fatty acid levels may negate the effects of chemotherapy. These recommendations have been adopted by the Dutch Cancer Society and by the Dutch National Working Group for Oncologic Dieticians. Increased Plasma Levels of Chemoresistance-Inducing Fatty Acid 16:4(n-3) After Consumption of Fish and Fish Oil. Daenen LGM, Cirkel GA, Houthuijzen JM, et al. JAMA Oncol. 2015;1:350-358
Fish oil has relevant levels of fatty acid 16:4(n-3) and preclinical models have shown that the fish oil neutralized the antitumor activity of chemotherapy, thus conferring drug resistance. With this preclinical information and given that cancer patients frequently use fish oil supplements, the authors evaluated the effect of fish oil intake in healthy volunteers, on the plasma levels of fatty acid 16:4(n-3), which has been shown to induce resistance to chemotherapeutic agents. The researchers first conducted a survey to determine what percentage of cancer patients undergoing treatment at a University Medical Center in the Netherlands were taking fish oil supplements. They also analyzed fatty acid 16:4(n-3) content, in 3 brands of fish oil supplements and 4 often consumed species of fish. The authors then randomly selected 30 healthy volunteers for the fish oil study and 20 healthy volunteers for the fish consumption study and the plasma levels of fatty acid 16:4(n-3) was measured after they consumed fish oil or fish, for a period of 2 weeks. They noted that 11% of the cancer patients in their study reported using omega-3 supplements. All fish oils tested contained amounts of fatty acid 16:4(n-3) ranging from 0.2 to 5.7 μM and this was adequate to induce chemoresistance to a variety of chemotherapeutic agents. They noted that there was a significant rise in the plasma 16:4(n-3) fatty acid levels in the healthy volunteers after they consumed fish oil supplements and fish, with high levels of fatty acid 16:4(n-3). Herring and Mackerel fish contained high levels of fatty acid 16:4(n-3), in contrast to Salmon and Tuna. The authors concluded that based on this preclinical data it is best to avoid fish oils and fish such as Herring and Mackerel in the 48 hours surrounding chemotherapy, as the high plasma 16:4(n-3) fatty acid levels may negate the effects of chemotherapy. These recommendations have been adopted by the Dutch Cancer Society and by the Dutch National Working Group for Oncologic Dieticians. Increased Plasma Levels of Chemoresistance-Inducing Fatty Acid 16:4(n-3) After Consumption of Fish and Fish Oil. Daenen LGM, Cirkel GA, Houthuijzen JM, et al. JAMA Oncol. 2015;1:350-358 This is accomplished by either surgical castration (bilateral orchiectomy) or medical castration using LHRH (GnRH- Gonadotropin-Releasing Hormone) agonists given along with 2 weeks of first generation anti-androgen agents such as EULEXIN® (Flutamide), CASODEX® (Bicalutamide) or NILANDRON® (Nilutamide), with the anti-androgen agents given to prevent testosterone flare. This large intergroup trial which was developed by the NCIC Clinical Trials Group in collaboration with the Medical Research Council and the National Cancer Institute US Cancer Therapy Evaluation Program, evaluated the benefits of adding Radiation Therapy (RT) to ADT, when compared to ADT alone, in patients with locally advanced prostate cancer. In this study, 1205 patients were randomly assigned to receive either ADT alone (N=602) or ADT plus RT (N=603). Eligible patients included those with T1-2 disease with either Prostate Specific Antigen (PSA) of more than 40 μg/L or PSA of 20-40 μg/L plus Gleason score of 8-10 or patients with T3-4, N0/NX, M0 prostate cancer. ADT consisted of either bilateral orchiectomy or LHRH agonists (plus 2 weeks of oral anti-androgen therapy to prevent testosterone flare), based on patient and physician preference, and ADT was continued for life. RT consisted of a dose of 64-69 Gy given in 35-39 fractions to the prostate gland and pelvis or prostate gland alone. The median age was 70 years and the median follow up was 8 years. Eighty seven percent of patients had T3-4 disease, 63% of patients had a PSA more than 20 μg/L and 18% had a Gleason score of more than 8. The Primary Endpoint was Overall Survival (OS), defined as the time from randomization to death from any cause. Secondary Endpoints included Time To Progression (TTP), improvement in Disease Specific Survival, quality of life and toxicity. The authors had previously reported the interim analysis findings of this intergroup trial and they noted that the addition of RT to ADT significantly improved overall survival, at a median follow up of 6 years (HR= 0.77; P=0.033). In this final analysis, at a median follow up of 8 years, the interim analysis findings were confirmed and the patients assigned to ADT plus RT had significantly improved Overall Survival compared to those who received ADT alone (HR=0.70; P<0.001), with a 30% reduction in the risk of death. Disease Specific Survival was also superior with ADT plus RT compared to ADT alone, with a 54% reduction in deaths from prostate cancer (HR=0.46; P <0 .001). There was a higher incidence of grade 1 and 2 bowel toxicities in patients who received ADT plus RT, but grade 3 bowel toxicities were rare and short term. The authors concluded that this long term follow up data suggests that the addition of Radiation Therapy to Androgen Deprivation Therapy significantly prolongs Overall and Disease Specific Survival, in patients with locally advanced prostate cancer. Final Report of the Intergroup Randomized Study of Combined Androgen-Deprivation Therapy Plus Radiotherapy Versus Androgen-Deprivation Therapy Alone in Locally Advanced Prostate Cancer. Mason MD, Parulekar WR, Sydes MR, et al. J Clin Oncol 2015; 33:2143-2150
This is accomplished by either surgical castration (bilateral orchiectomy) or medical castration using LHRH (GnRH- Gonadotropin-Releasing Hormone) agonists given along with 2 weeks of first generation anti-androgen agents such as EULEXIN® (Flutamide), CASODEX® (Bicalutamide) or NILANDRON® (Nilutamide), with the anti-androgen agents given to prevent testosterone flare. This large intergroup trial which was developed by the NCIC Clinical Trials Group in collaboration with the Medical Research Council and the National Cancer Institute US Cancer Therapy Evaluation Program, evaluated the benefits of adding Radiation Therapy (RT) to ADT, when compared to ADT alone, in patients with locally advanced prostate cancer. In this study, 1205 patients were randomly assigned to receive either ADT alone (N=602) or ADT plus RT (N=603). Eligible patients included those with T1-2 disease with either Prostate Specific Antigen (PSA) of more than 40 μg/L or PSA of 20-40 μg/L plus Gleason score of 8-10 or patients with T3-4, N0/NX, M0 prostate cancer. ADT consisted of either bilateral orchiectomy or LHRH agonists (plus 2 weeks of oral anti-androgen therapy to prevent testosterone flare), based on patient and physician preference, and ADT was continued for life. RT consisted of a dose of 64-69 Gy given in 35-39 fractions to the prostate gland and pelvis or prostate gland alone. The median age was 70 years and the median follow up was 8 years. Eighty seven percent of patients had T3-4 disease, 63% of patients had a PSA more than 20 μg/L and 18% had a Gleason score of more than 8. The Primary Endpoint was Overall Survival (OS), defined as the time from randomization to death from any cause. Secondary Endpoints included Time To Progression (TTP), improvement in Disease Specific Survival, quality of life and toxicity. The authors had previously reported the interim analysis findings of this intergroup trial and they noted that the addition of RT to ADT significantly improved overall survival, at a median follow up of 6 years (HR= 0.77; P=0.033). In this final analysis, at a median follow up of 8 years, the interim analysis findings were confirmed and the patients assigned to ADT plus RT had significantly improved Overall Survival compared to those who received ADT alone (HR=0.70; P<0.001), with a 30% reduction in the risk of death. Disease Specific Survival was also superior with ADT plus RT compared to ADT alone, with a 54% reduction in deaths from prostate cancer (HR=0.46; P <0 .001). There was a higher incidence of grade 1 and 2 bowel toxicities in patients who received ADT plus RT, but grade 3 bowel toxicities were rare and short term. The authors concluded that this long term follow up data suggests that the addition of Radiation Therapy to Androgen Deprivation Therapy significantly prolongs Overall and Disease Specific Survival, in patients with locally advanced prostate cancer. Final Report of the Intergroup Randomized Study of Combined Androgen-Deprivation Therapy Plus Radiotherapy Versus Androgen-Deprivation Therapy Alone in Locally Advanced Prostate Cancer. Mason MD, Parulekar WR, Sydes MR, et al. J Clin Oncol 2015; 33:2143-2150