
The FDA on September 13, 2016 modified the dosage regimen for OPDIVO®, which is currently approved for Renal Cell Carcinoma, metastatic Melanoma, and Non Small Cell Lung Cancer. The currently approved recommended dosage regimens were modified to 240 mg intravenously (IV) every two weeks. OPDIVO® is marketed by Bristol-Myers Squibb company.
Author: RR
Postmastectomy Radiotherapy ASCO Guideline Update
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 246,660 new cases of invasive breast cancer will be diagnosed in 2016 and 40,450 women will die of the disease. The update of the American Society of Clinical Oncology guideline concerning use of PostMastectomy RadioTherapy (PMRT) published in 2001, was developed by an expert panel following review of relevant literature published between January 2001 and July 2015 and further included a meta-analysis of 22 clinical trials published in 2014. Even though the use of PMRT has been widely accepted for patients with four or more positive lymph nodes, the role of PMRT for those with one to three positive nodes still remains controversial. This update addresses the issues at large and provides guidelines to help Health Care Providers and patients make informed decisions.
Clinical Question 1
Is PMRT indicated in patients with T1-2 tumors with one to three positive axillary lymph nodes who undergo Axillary Lymph Node Dissection (ALND)?
Recommendation 1a: The panel unanimously agreed that the available evidence shows that PMRT reduces the risks of locoregional failure, any recurrence, and breast cancer mortality for patients with T1-2 breast cancer and one to three positive lymph nodes. However, one has to weigh the risk and benefit with PMRT and individualize therapy. Patients with low tumor burden, favorable tumor characteristics, comorbidities, or coexisting conditions and limited life expectancy, may not be appropriate candidates for PMRT, as its potential toxicities outweigh the absolute benefit of PMRT.
Recommendation 1b: The decision to use PMRT should be made in a multidisciplinary fashion through discussion among providers from all treating disciplines, early in a patient’s treatment course, soon after surgery or before or soon after the initiation of systemic therapy.
Recommendation 1c: Decision making must fully involve the patients, so that they are able to weigh the risk/benefits of PMRT, with the best information provided by the treating Health Care Provider.
Clinical Question 2
Is PMRT indicated in patients with T1-2 tumors and a positive Sentinel Node Biopsy (SNB) who do not undergo completion ALND?
Recommendation: Patients with T1-2 tumors with positive sentinel lymph node biopsy, who choose not to have Axillary Lymph Node Disection, should receive PMRT only if there is already sufficient information to justify its use, without needing to know that additional axillary nodes are involved. SNB is generally performed at the time of mastectomy for this patient group, with omission of ALND if the nodes are negative. If the sentinel nodes are positive, ALND is performed. There is increasing controversy about whether ALND is always necessary, if there is limited disease in the affected sentinel nodes. This practice is based on extrapolation of data from randomized trials of patients treated exclusively or predominantly with breast-conserving surgery and whole-breast irradiation or breast plus axillary irradiation.
Clinical Question 3
Is PMRT indicated in patients presenting with clinical stage I or II cancers who have received NeoAdjuvant Systemic Therapy (NAST)?
Recommendation: Patients with axillary nodal involvement that persists after NAST, such as less than a complete pathologic response, should receive PMRT. Observational data suggest a low risk of locoregional recurrence for patients who have clinically negative nodes and receive NAST or who have a complete pathologic response in the lymph nodes with NAST.
Clinical Question 4
Should Regional Nodal Irradiation (RNI) include both the Internal Mammary (IMNs) and supraclavicular-axillary apical nodes when PMRT is used in patients with T1-2 tumors with one to three positive axillary nodes?
Recommendation: Radiation Therapy should generally be administered to both the IMNs and the supraclavicular-axillary apical nodes in addition to the chest wall or reconstructed breast, when PMRT is used for patients with positive axillary lymph nodes.
Postmastectomy Radiotherapy: An American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology Focused Guideline Update. Recht A, Comen EA, Fine RE, et al. Published online before print September 19, 2016, doi:10.1200/JCO.2016.69.1188
DARZALEX® Combination Demonstrates Impressive Efficacy in Relapsed or Refractory Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, about 30,330 new cases will be diagnosed in 2016 and 12,650 patients will die of the disease. Multiple Myeloma is a disease of the elderly, with a median age at diagnosis of 69 years and characterized by intrinsic clonal heterogeneity. With a record number of regulatory approvals for Myeloma treatment over the past 12 years, the median survival for patients with Myeloma is over 10 years. The recent new drugs approved for the treatment of relapsed/refractory Multiple Myeloma include a Histone Decetylase inhibitor (FARYDAK®) and 2 monoclonal antibodies, Daratumumab (DARZALEX®) and Elotuzumab (EMPLICITI®).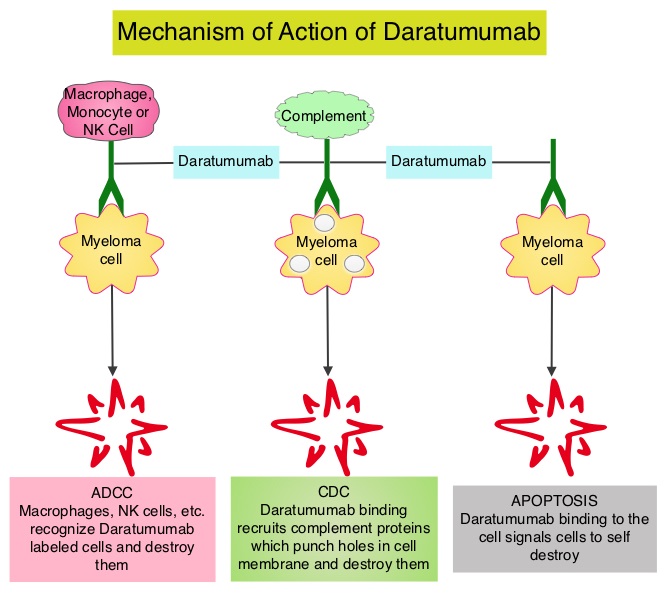
Daratumumab (DARZALEX®) is a human IgG1 antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Mediated Cytotoxicity and direct apoptosis. Additionally, DARZALEX® may have a role in immunomodulation by depleting CD38-positive regulator Immune suppressor cells, and thereby expanding T cells, in patients responding to therapy. Previously published phase I and II studies involving patients with relapsed or refractory multiple myeloma demonstrated promising efficacy of DARZALEX® when given as a single agent, as well as when given along with Lenalidomide (REVLIMID®) and Dexamethasone.
The authors now report the results of a prespecified interim analysis of a phase III trial of DARZALEX®, REVLIMID® and Dexamethasone in patients with relapsed or refractory multiple myeloma. In this randomized, open-label, multicenter, phase III study, 569 patients who had relapsed or refractory multiple myeloma were assigned in a 1:1 ratio to receive either DARZALEX®, REVLIMID® and Dexamethasone (Daratumumab group, N=286) or REVLIMID® and Dexamethasone (Control group, N=283). Patients refractory to REVLIMID® were excluded. Patients in the Daratumumab group received DARZALEX® 16 mg/kg IV on days 1, 8, 15, and 22 of a 28 day cycle for 8 weeks during cycles 1 and 2, every 2 weeks (on days 1 and 15) for 16 weeks (cycles 3 thru 6), and every 4 weeks thereafter. Both treatment groups received REVLIMID® 25 mg PO on days 1-21 of each cycle and Dexamethasone 40 mg PO weekly. The primary end point was Progression Free Survival (PFS). Secondary end points included the time to disease progression, Response Rate, time to response, duration of response, Overall Survival (OS) and percentage of patients with results below the threshold for Minimal Residual Disease. Minimal Residual Disease status was evaluated for patients who had a Complete Response by Next-Generation sequencing assay of bone marrow.
At a median follow-up of 13.5 months, the PFS at 12 months was 83.2% in the Daratumumab group compared to 60.1% in the control group (HR=0.37; P<0.001). The Overall Response Rate was significantly higher in the Daratumumab group than in the control group (92.9% versus 76.4%, P<0.001) and further, there was a higher rate of Complete Response or better (43.1% vs. 19.2%, P<0.001). In the Daratumumab group, 22.4% of the patients had results below the threshold for Minimal Residual Disease (1 tumor cell per 105 white cells), as compared with 4.6% of those in the control group (P<0.001), and those with results below the threshold for Minimal Residual Disease had improved outcomes. The most common grade 3 or 4 adverse events were cytopenias. Approximately 48% of the patients in the Daratumumab group experienced grade 1 or 2 infusion-related reactions.
The authors concluded that the addition of DARZALEX® to REVLIMID® and Dexamathasone significantly improves Progression Free Survival among patients with relapsed or refractory multiple myeloma. This impressive efficacy data may warrant the use of this triplet combination for first relapse, in this group of patients, provided their disease is not refractory to REVLIMID®. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. Dimopoulos MA, Oriol A, Nahi H, et al. for the POLLUX Investigators. N Engl J Med 2016;375:1319-1331
CDK4 and CDK6 Inhibitor ABEMACICLIB Highly Effective in Refractory Hormone Receptor Positive Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 246,660 new cases of invasive breast cancer will be diagnosed in 2016 and 40,450 women will die of the disease. Estrogen Receptor (ER) positive breast cancer cells are driven by estrogens. Approximately 80% of breast tumors express Estrogen Receptors and/or Progesterone Receptors and these patients are often treated with anti-estrogen therapy as first line treatment. Cyclin Dependent Kinases (CDK) play a very important role to facilitate orderly and controlled progression of the cell cycle. Genetic alterations in these kinases and their regulatory proteins have been implicated in various malignancies. Cyclin Dependent Kinases 4 and 6 (CDK4 and CDK6), phosphorylate RetinoBlastoma protein (RB), and initiate transition from the G1 phase to the S phase of the cell cycle. CDK4 and CDK6 are activated in hormone receptor positive breast cancer, promoting breast cancer cell proliferation. Further, there is evidence to suggest that endocrine resistant breast cancer cell lines depend on CDK4 for cell proliferation.
Abemaciclib is an oral, selective inhibitor of CDK4 and CDK6 kinase activity that prevents the phosphorylation and subsequent inactivation of the Rb tumor suppressor protein, thereby inducing G1 cell cycle arrest and inhibition of cell proliferation. The FDA granted breakthrough designation for Abemaciclib based on a phase I trial in which this agent demonstrated significant single agent activity in refractory Hormone Receptor (HR) positive metastatic breast cancer. MONARCH 1 is a single arm phase II study which evaluated the single-agent activity of Abemaciclib in heavily pretreated patients with HR-positive, HER2-negative metastatic breast cancer. This trial included 132 patients with HR-positive, HER2-negative metastatic breast cancer, whose disease progressed on or after endocrine therapy and chemotherapy. Patients had received a median of 3 lines of prior therapy for advanced disease and this included a median of 2 lines of chemotherapy. Approximately 50% of the patients had received FASLODEX® (Fulvestrant), 70% of patients had received Taxane based chemotherapy and 55 % of patients had received XELODA® (Capecitabine), in the metastatic setting. Abemaciclib was administered at 200 mg PO daily on a continuous schedule every 12 hours until disease progression. The median age was 58 yrs, 90% of the patients had visceral disease and 85% had at least 2 sites of metastatic disease. The primary endpoint was Objective Response Rate (ORR) and secondary endpoints included Duration of Response, Progression Free Survival (PFS), Overall Survival (OS), Clinical Benefit Rate (Complete Response plus Partial Response plus Stable Disease) and safety.
An interim analysis was performed at 8 months by when 35.6% of patients had received at least 8 cycles of Abemaciclib. The ORR was 17.4%, the Clinical Benefit Rate lasting for 6 months or more was 42.4%. The median time to response was 3.7 months and the median Duration of Response was 8.6 months. The median PFS was 5.7 months. The most common adverse events were diarrhea, fatigue, nausea, decreased appetite, abdominal pain and treatment discontinuation rate was infrequent at 6.8%.
It was concluded that CDK4 and CDK6 inhibitor, Abemaciclib, has significant antitumor activity in patients with refractory, HR-positive, HER2-negative metastatic breast cancer, for whom chemotherapy is the only option. Studies are underway combining Abemaciclib with FASLODEX®, for postmenopausal patients with HR-positive, HER2-negative, locally advanced or metastatic breast cancer who had progressed on 1 prior endocrine therapy (MONARCH-2 trial), as well as combining Abemaciclib with a nonsteroidal Aromatase Inhibitor in an earlier setting (MONARCH-3 trial), for patients with HR-positive, HER2-negative, locoregionally recurrent or metastatic breast cancer. MONARCH 1: Results from a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as monotherapy, in patients with HR+/HER2- breast cancer, after chemotherapy for advanced disease. Dickler MN, Tolaney SM, Rugo HS, et al. J Clin Oncol 34, 2016 (suppl; abstr 510)
Adjuvant Therapy with YERVOY® Improves Overall Survival in Stage III Melanoma
SUMMARY: It is estimated that in the US, approximately 76,380 new cases of melanoma will be diagnosed in 2016 and approximately 10,130 patients will die of the disease. The incidence of melanoma has been on the rise for the past three decades. Stage III malignant melanoma is a heterogeneous disease and the risk of recurrence is dependent on the number of positive nodes as well as presence of palpable versus microscopic nodal disease. Further, patients with a metastatic focus of more than 1 mm in the greatest dimension in the affected lymph node, have a significantly higher risk of recurrence or death than those with a metastasis of 1 mm or less. Patients with stage IIIA disease have a disease-specific survival rate of 78%, whereas those with stage IIIB and stage IIIC disease have a disease-specific survival rates of 59% and 40% respectively. Immune checkpoints are cell surface inhibitory proteins/receptors that harness the immune system and prevent uncontrolled immune reactions. With the recognition of Immune checkpoint proteins (“gate keepers”) and their role in suppressing antitumor immunity, antibodies have been developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By blocking the Immune checkpoint proteins, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response.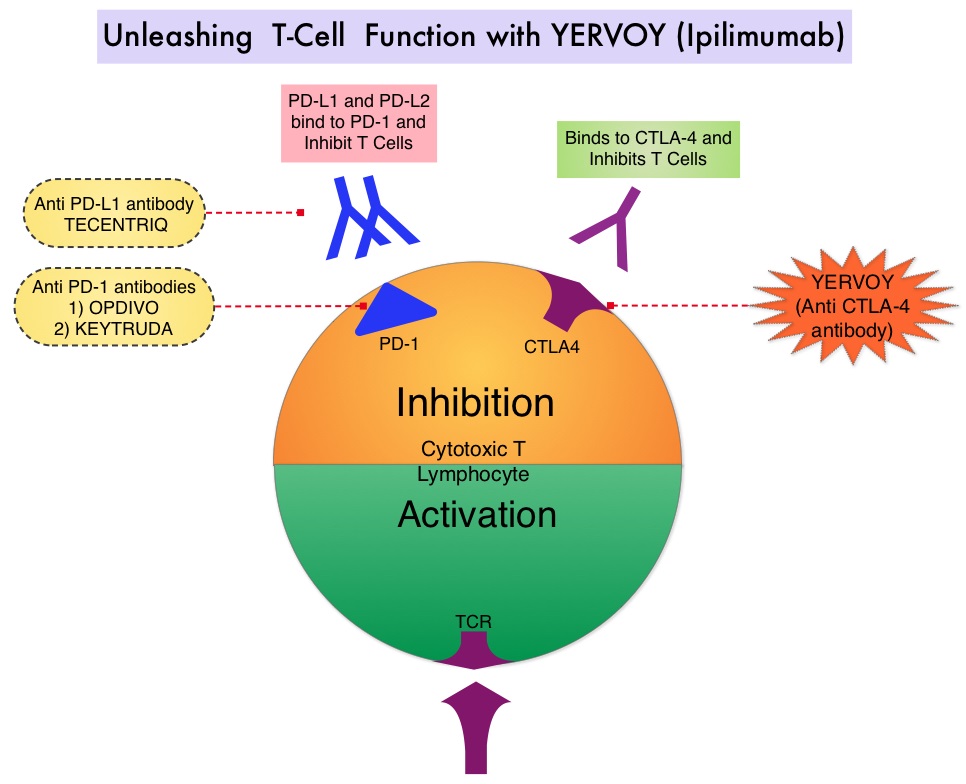
YERVOY® is a fully human immunoglobulin G1 monoclonal antibody, that blocks Immune checkpoint protein/receptor CTLA-4. YERVOY® was approved by the FDA in 2015 as adjuvant therapy for patients with malignant melanoma based on a phase III trial (EORTC 18071) in which adjuvant YERVOY® was compared with placebo in patients with resected stage III melanoma. At a median follow-up of 2.7 years, adjuvant YERVOY® was associated with significantly prolonged Recurrence Free Survival compared with placebo (HR=0.75; P=0.001). In this study, YERVOY® was dosed at 10 mg/kg and this dose was chosen based on a phase II trial that evaluated YERVOY® administered at three dose levels – 0.3 mg/kg, 3 mg/kg and 10 mg/kg. This trial demonstrated that the 10 mg/kg dose had the greatest efficacy for treatment of advanced melanoma, but had more toxicity.
The authors have now reported the efficacy of adjuvant therapy with YERVOY® in patients with high-risk stage III melanoma after complete lymph node dissection, at a median follow up of 5.3 years. EORTC 18071 is a randomized, double-blind, phase III trial in which patients who had undergone complete resection of stage III cutaneous melanoma were randomly assigned in a 1:1 ratio to receive YERVOY® at a dose of 10 mg/kg (N=475) or placebo (N=476), every 3 weeks for four doses and then every 3 months for up to 3 years or until disease recurrence or unacceptable toxicities. The Primary end point was Recurrence Free Survival and Secondary end points included Overall Survival, distant metastasis-free survival and safety.
At a median follow up of 5.3 years, the 5-year Recurrence Free Survival was 40.8% in the YERVOY® group compared with 30.3% in the placebo group (HR for recurrence or death = 0.76; P<0.001) and the 5-year Overall Survival was 65.4% in the YERVOY® group compared with 54.4% in the placebo group (HR for death = 0.72; P=0.001). The later meant a 28% reduction in the risk of death. The distant metastasis-free survival rate at 5 years was 48.3% in the YERVOY® group compared with 38.9% in the placebo group (HR for death or distant metastasis = 0.76; P=0.002). Grade 3 or 4 adverse events occurred in 54.1% of the patients in the YERVOY® group compared to 26.2% in the placebo group, and this was significant. Grade 3 or 4 Immune-related adverse events occurred in 41.6% of the patients in the YERVOY® group compared to 2.7% in the placebo group.
It was concluded that adjuvant therapy with YERVOY® at a dose of 10 mg/kg for high-risk stage III melanoma, significantly improves Recurrence Free Survival, Overall Survival, and distant metastasis-free survival, when compared to placebo. Given the potentially severe toxicities with this dose level of YERVOY®, the risks and benefits of this treatment has to be discussed with the patients and this treatment option should be reserved for experienced centers. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. Eggermont AM, Chiarion-Sileni V, Grob J-J, et al. October 8, 2016DOI: 10.1056/NEJMoa1611299
ASCO Guidelines on Adjuvant Targeted Therapy for HER2-Positive Breast Cancers
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their life time. Approximately, 246,660 new cases of invasive breast cancer will be diagnosed in 2016 and 40,450 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 20-25% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. HERCEPTIN® (Trastuzumab) is a humanized monoclonal antibody targeting HER2. HERCEPTIN® binds to subdomain IV of the HER2 extracellular domain and blocks the downstream cell signaling pathways (PI3K-AKT pathway) and induces Antibody Dependent Cellular Cytotoxicity (ADCC). Adjuvant chemotherapy in combination with HERCEPTIN® has been shown to reduce the relative risk of relapse by 52% and relative risk of death by 33%. ASCO has established a process for adapting clinical practice guidelines of other organizations and this summary of the practice recommendations were adapted from the Cancer Care Ontario evidence based clinical practice guidelines, for the adjuvant treatment of HER2-positive early breast cancers.
Guideline Question: What is the optimal use of cytotoxic chemotherapy and Human Epidermal growth factor Receptor 2 (HER2) – directed therapy?
Target Population: Female patients who are being considered for, or who are receiving, systemic therapy after definitive surgery for early invasive breast cancer, defined largely as invasive cancer stages I to IIA (T1N0-1, T2N0).
RECOMMENDATIONS
Use of an Anthracycline-Taxane Regimen
1) An adjuvant chemotherapy regimen containing Anthracycline-Taxane is considered the optimal strategy for high risk patients, if they are able to tolerate this regimen.
2) For patients with high-risk disease who will not receive a Taxane, an optimal-dose Anthracycline three-drug regimen (cumulative dose of doxorubicin ≥ 240 mg/m2 or epirubicin ≥ 600 but no higher than 720 mg/m2) that contains Cyclophosphamide, is recommended. The cumulative dose of Doxorubicin in two-drug regimens should not exceed 240 mg/m2.
3) The addition of Gemcitabine or Capecitabine to an Anthracycline-Taxane regimen is not recommended for adjuvant chemotherapy.
Capecitabine in Patients 65 Years of Age and Older
In patients age 65 years or older, Capecitabine is not recommended as an adjuvant chemotherapy option in lieu of standard regimens like Doxorubicin plus Cyclophosphamide (AC) or Cyclophosphamide, Methotrexate, and Fluorouracil (CMF with oral cyclophosphamide).
CMF as an Alternative to AC
For patients in whom Anthracycline-Taxane is contraindicated, CMF (with oral cyclophosphamide) is an acceptable chemotherapy alternative to AC. The ASCO panel recommends classic CMF (oral cyclophosphamide days 1 to 14 with IV Methotrexate-Fluorouracil days 1 and 8, repeated every 28 days for six cycles) as the default adjuvant CMF regimen. However, the panel also recognizes IV CMF regimen given every 21 days.
Acceptable Adjuvant Chemotherapy Regimens for Patients with Higher-Risk Early-Stage Breast Cancer
1) FEC (Fluorouracil, Epirubicin, and Cyclophosphamide) × 3 → T (Docetaxel) × 3 (superior to FEC × 6)
2) AC × 4 → T × 4 (superior to AC × 4)
3) Docetaxel, Doxorubicin, and Cyclophosphamide × 6 (superior to Fluorouracil, Doxorubicin, and Cyclophosphamide × 6)
4) AC × 4 → paclitaxel (P) administered weekly
5) Dose-Dense AC → P (every 2 weeks)
Adjuvant Regimen When an Anthracycline Is Not Preferred
1) Docetaxel plus Cyclophosphamide (TC) × 4 is recommended as an alternative to AC × 4
2) Classic CMF with oral Cyclophosphamide for six cycles. The panel also recognizes IV CMF regimen given every 21 days.
Patient Selection and Adjuvant Trastuzumab Therapy
Only patients with HER2-positive breast cancer (overexpressed based on ImmunoHistoChemistry (IHC 3+) or amplified based on in situ hybridization [ISH ratio ≥ 2.0 or average HER2 copy number ≥ 6.0]), should be offered adjuvant Trastuzumab.
Trastuzumab Plus Chemotherapy
1) Trastuzumab plus chemotherapy is recommended for all patients with HER2-positive, node-positive breast cancer and for patients with HER2-positive, node-negative breast cancer greater than 1 cm in size.
2) Trastuzumab therapy can be considered in small, node-negative tumors (1 cm or less).
3) Trastuzumab can be administered with any acceptable adjuvant chemotherapy regimen.
4) The administration of Trastuzumab concurrently with the Anthracycline component of a chemotherapy regimen is not recommended because of the potential for increased cardiotoxicity.
5) Trastuzumab should be preferentially administered concurrently (not sequentially) with a non-Anthracycline chemotherapy regimen.
6) Less cardiotoxicity is seen with TCH (Docetaxel, Carboplatin, and Trastuzumab) than with AC→TH (Doxorubicin and Cyclophosphamide→Docetaxel and Trastuzumab), and TCH is recommended for patients at higher risk for cardiotoxicity.
7) Even though there is no phase III evidence for the addition of Trastuzumab to some chemotherapy regimens, such as TC, those regimens might be in use and are reasonable options, particularly to mitigate cardiotoxicity in certain patients.
Duration of Trastuzumab Therapy and Cardiac Function Assessment
Patients should be offered 1 year total of adjuvant Trastuzumab, with regular assessments of cardiac function during that period.
Selection of Optimal Adjuvant Chemotherapy Regimens for Early Breast Cancer and Adjuvant Targeted Therapy for HER2-Positive Breast Cancers: An American Society of Clinical Oncology Guideline Adaptation of the Cancer Care Ontario Clinical Practice Guideline. Denduluri N, Somerfield MR, Eisen A, et al. J Clin Oncol 2016;34:2416-2427
FDA Approves TAGRISSO® (Osimertinib) Blood-Based T790M Companion Diagnostic Test
SUMMARY: The FDA on September 29, 2016 approved a blood-based companion diagnostic for TAGRISSO® (Osimertinib). The companion diagnostic for TAGRISSO® is the only FDA approved and clinically validated companion diagnostic test that uses either tissue or a blood sample to confirm the presence of a T790M point mutation in patients with metastatic Epidermal Growth Factor Receptor (EGFR) mutation-positive Non Small Cell Lung Cancer (NSCLC), who have had progression of disease on or after EGFR Tyrosine Kinase Inhibitor therapy. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2016 about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. Approximately 10% to 15% of Caucasian patients and 50% of Asian patients with Adenocarcinomas, harbor activating EGFR mutations and 90% of these mutations are either Exon 19 deletions or L858R point mutations in Exon 21. EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60% to 70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9 to 14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50% – 60% of patients.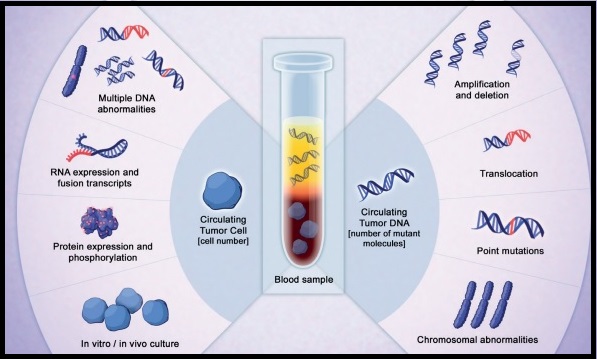
TAGRISSO® is presently approved by the FDA for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, who had progressed on prior systemic therapy, including an EGFR TKI. The application of precision medicine with targeted therapy requires detection of molecular abnormalities in a tumor specimen, following progression or recurrence. Archived biopsy specimens may not be helpful as it is important to identify additional mutations in the tumor at the time of recurrence or progression, in order to plan appropriate therapy. Further, recurrent tumors may be inaccessible for a safe biopsy procedure or the clinical condition of the patient may not permit a repeat biopsy. Additionally, the biopsy itself may be subject to sampling error due to tumor heterogeneity. Genotyping circulating-free tumor DNA (cfDNA) in the plasma can potentially overcome the shortcomings of repeat biopsies and tissue genotyping, allowing the detection of many more targetable gene mutations, thus resulting in better evaluation of the tumor genome landscape.
The COBAS® Mutation Test v2, is a real-time PCR test for the qualitative detection of defined mutations of the EGFR gene in NSCLC patients. Defined EGFR mutations are detected using DNA isolated from Formalin-Fixed Paraffin-Embedded Tumor tissue (FFPET) or circulating-free tumor DNA (cfDNA) from plasma, obtained from EDTA anti-coagulated peripheral whole blood (purple top tube). This new blood-based companion diagnostic test offers an important option to identify T790M mutation in patients with metastatic EGFR mutation-positive NSCLC, who have progressed on an EGFR TKI therapy, and for whom a tissue biopsy may not be feasible.
US FDA approves Tagrisso (osimertinib) blood-based T790M companion diagnostic test. AstraZeneca website. https://www.astrazeneca-us.com/content/az-us/media/press-releases/2016/us-fda-approves-tagrisso-osimertinib-blood-based-t790m-companion-diagnostic-test-09292016.html. Updated September 29, 2016.
MammaPrint® Identifies Women with Early Stage Breast Cancer Who Can Avoid Adjuvant Chemotherapy
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 246,660 new cases of invasive breast cancer will be diagnosed in 2016 and 40,450 women will die of the disease. Patients with early stage breast cancer often receive adjuvant chemotherapy. Chemotherapy recommendations for early stage breast cancer are often made based on tumor size, grade, hormone receptor and HER-2 status, immunohistochemical markers such as Ki-67, nodal status, patients age, menopausal and performance status. Adjuvant! Online is one of the tools that incorporates these features and assists in treatment decision making. This tool however does not take into account individual tumor molecular signatures which can better predict clinical outcomes. MammaPrint® is a 70-gene signature assay approved by the FDA and is able to distinguish low risk and high risk tumors based on the risk of distant recurrence at 5 and 10 years. The authors in this study selected patients for adjuvant chemotherapy utilizing 70-gene signature assay in addition to standard clinicopathological criteria and prospectively reported 5-year outcomes in the treatment groups.
MINDACT (Microarray in Node-Negative and 1 to 3 Positive Lymph Node Disease May Avoid Chemotherapy) study is a international, prospective, randomized, phase III study which enrolled 6693 women with early-stage breast cancer and determined their genomic risk using the 70-gene signature assay (MammaPrint®) and their clinical risk using a modified version of Adjuvant! Online. Enrolled patients were divided into four groups based on their clinical and genomic risk: low clinical risk and low genomic risk (N=2745, 41%); high clinical risk and high genomic risk (N=1806, 27%), high clinical risk and low genomic risk (N=1550, 23.2%); low clinical risk and high genomic risk (N=592, 8.8%). Patients in the first 2 concordant groups were treated according to their risk category, ie. low-risk group did not receive adjuvant chemotherapy whereas adjuvant chemotherapy was added to endocrine therapy following surgery, in the high-risk patient group. Patients in the last 2 discordant groups were randomly assigned to receive adjuvant chemotherapy or no chemotherapy. The median age of the patients was 55 yrs, 79% of the patients had node-negative disease and 21% had 1-3 positive nodes. A total of 88% of the tumors expressed ER, PR, or both, and 9.5% were HER-2 positive.
The primary endpoint of this study was to show noninferiority against a predefined benchmark of a 5-year metastasis-free survival rate of 92%, in patients at high clinical risk, for whom a discordant low genomic risk led to the omission of otherwise standard adjuvant chemotherapy. It was noted that in this group of patients at high clinical risk and low genomic risk (N=1550) at 5 years, the rate of survival without distant metastasis was 94.7% among those not receiving chemotherapy, and this met the study criterion for noninferiority, with similar outcomes noted in all sub groups of patients.. The absolute difference in the survival rate between those who did not receive adjuvant chemotherapy and those who received chemotherapy was 1.5%, with the survival rate slightly higher in those who received adjuvant chemotherapy. The 5-year metastasis-free survival rate for women who were low risk by both genomic and clinical criteria and who did not receive adjuvant chemotherapy was 97.6%, compared with 90.6% for those women who were high risk by both criteria and who did receive adjuvant chemotherapy.
It was concluded that in patients with early stage breast cancer, who were considered to be at high clinical risk for recurrence, 70-gene signature assay was able to identify those with low genomic risk, who had marginal benefit with adjuvant chemotherapy. With treatment guidance using 70-gene signature assay, approximately 46% of early stage breast cancer patients with high clinical risk and low genomic risk might not require adjuvant chemotherapy. 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. Cardoso F, van’t Veer LJ, Bogaerts J, et al. N Engl J Med 2016; 375:717-729
Location of Primary Tumor Predicts Survival and Choice of Treatment in Metastatic Colorectal Cancer
SUMMARY: ColoRectal Cancer (CRC) is the third most common cancer diagnosed in both men and women in the United States. The American Cancer Society estimates that approximately 135,000 new cases of ColoRectal Cancer will be diagnosed in the United States in 2016 and over 49,000 patients are expected to die of the disease. Even though prognosis for patients with Colon Cancer has improved over the past two to three decades, it has remained unclear if improvement in survival was greater for left sided Colon Cancer versus cancer originating in the Right hemicolon.
Venook and colleagues had previously presented data from the primary analysis of a phase III CALGB/SWOG 80405 study which compared AVASTIN®: (Bevacizumab) or ERBITUX® (Cetuximab), given in conjunction with FOLFOX (Leucovorin, 5-Fluorouracil, Oxaliplatin) or FOLFIRI (Leucovorin, 5-FU, Irinotecan), as first line treatment of metastatic colorectal cancer. This study showed that chemotherapy plus ERBITUX® resulted in similar Overall Survival (29.9 months) as chemotherapy plus AVASTIN® (29 months) as well as Progression Free Survival (PFS). The present publication is a retrospective evaluation from the phase III 80405 clinical trial which included data from 1,025 patients with KRAS wild-type disease. The researchers assessed the impact of tumor location on Overall and PFS in this group of patients. The median age was 59 yrs and 293 patients had a right-sided primary tumor (location in the cecum to hepatic flexure) and 732 patients had a left-sided primary tumor (location in the splenic flexure to rectum).
It was noted that patients with tumors originating in the right side of the colon had much shorter median Overall Survival (19.4 months) compared to patients with left-sided tumors (33.3 months), (HR=1.60; P<0.001), with a 14 month survival improvement in the left versus right-sided primary tumors, for patients with metastatic disease. Tumor location in the colon also had a bearing on response to ERBITUX® and AVASTIN®. For the patient group who received ERBITUX®, the median Overall Survival (OS) was 36 months for patients with left-sided tumors but 16.7 months for patients with right-sided tumors. For those who received AVASTIN®, the median OS was 31.4 months for patients with left-sided tumors and 24.2 months for those with right-sided tumors. The median PFS was also influenced by the site of the primary tumor and was 8.9 months for right-sided tumors versus 11.5 months for left-sided tumors in the overall cohort of patients (HR = 1.26; P = 0.002). In a separate analysis, the authors also evaluated data from an additional 213 patients in CALGB/SWOG 80405 study who harbored KRAS mutations. These patients were originally included in this study prior to knowledge that ERBITUX® only benefited KRAS wild-type tumors. In this group of patients, those with left-sided primary tumors again achieved a longer median OS (30.3 months) compared to 23.1 months, among patients with right-sided tumors.
It was concluded that the biology of tumors originating in the right colon may be different from those originating in the left colon with ERBITUX® showing superiority over AVASTIN® when combined with chemotherapy in KRAS wild-type patients with left-sided colon cancer, whereas patients with right-sided colon cancer appear to benefit more from AVASTIN® based chemotherapy regimen. Regardless of KRAS mutational status, AVASTIN® based, first line chemotherapy regimen should be considered, for all patients with metastatic colorectal cancer, with a right-sided primary tumor. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). Venook AP, Niedzwiecki D, Innocenti F, et al. J Clin Oncol 34, 2016 (suppl; abstr 3504)
Andexanet Alfa – An Antidote for Acute Major Bleeding Associated with New Oral Anticoagulants
SUMMARY: There are presently four New Oral Anticoagulants approved in the United States for the treatment of Venous ThromboEmbolism. They include PRADAXA® (Dabigatran), which is a direct thrombin inhibitor and XARELTO® (Rivaroxaban), ELIQUIS® (Apixaban), SAVAYSA® (Endoxaban), which are Factor Xa inhibitors. Compared to COUMADIN® (Warfarin), the New Oral Anticoagulants have a rapid onset of action, wider therapeutic window, shorter half-lives (7-14 hours in healthy individuals), no laboratory monitoring and fixed dosing schedule. The half life of these agents can however be prolonged in those with renal insufficiency and may be unsafe and direct oral anticoagulants are ineffective in patients with mechanical heart valves. In several clinical studies, these New Oral Anticoagulants have been shown to reduce the rate of major bleeding by 28% and the rates of intracranial and fatal hemorrhage by 50%, when compared to COUMADIN®. Unlike bleeding caused by COUMADIN®, which can be reversed using Vitamin K or Fresh Frozen Plasma, there are no specific agents presently available, for reversing bleeding caused by the New Oral Anticoagulants or for stopping the anticoagulant effects of these drugs, in patients who need urgent surgical intervention. The FDA in October 16, 2015, granted accelerated approval to Idarucizumab (PRAXBIND®), for the treatment of patients treated with Dabigatran (PRADAXA®), a direct thrombin inhibitor, when reversal of the anticoagulant effects of PRADAXA® is needed for emergency surgery/urgent procedures, or in life-threatening or uncontrolled bleeding. However, the other New Oral Anticoagulants approved in the United States for the treatment of Venous ThromboEmbolism such as XARELTO® (Rivaroxaban), ELIQUIS® (Apixaban), SAVAYSA® (Endoxaban), are Factor Xa inhibitors and do not have an antidote. As such, some Health Care Providers have discouraged their patients from taking these direct oral anticoagulants until an antidote became available, should their patients need urgent surgical intervention.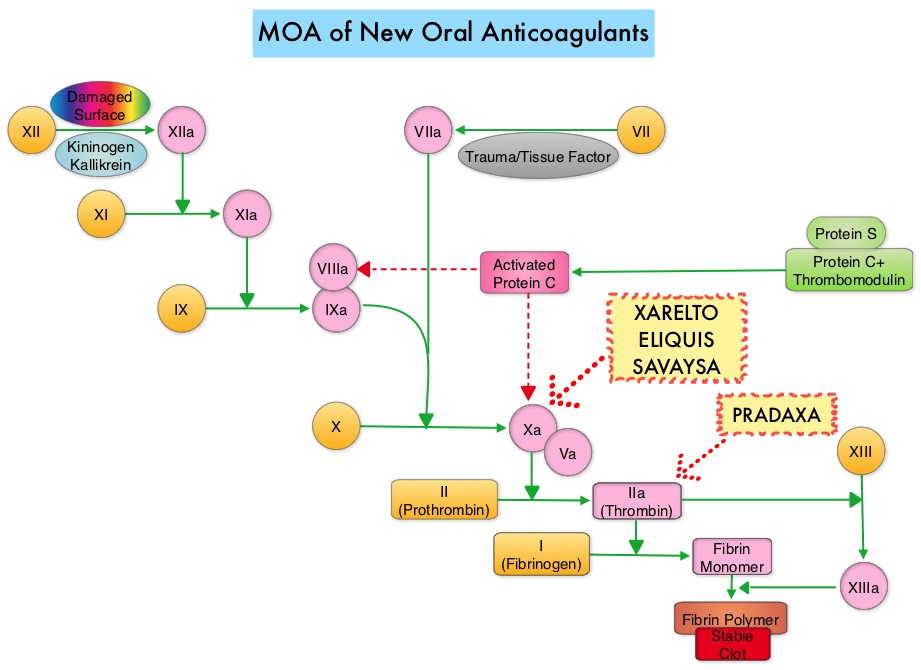
Andexanet alfa (AndexXa®) is a recombinant, modified human Factor Xa decoy protein without intrinsic catalytic activity, that binds Factor Xa inhibitors. In a previously published study, AndexXa® reversed the anticoagulant activity of ELIQUIS® and XARELTO® in older healthy participants within minutes after administration, and reduced both the unbound fraction of the plasma level of factor Xa inhibitor and anti-Factor Xa activity, with minimal clinical toxicity (N Engl J Med 2015; 373:2413-242). The AndexXa®, a Novel Antidote to the Anticoagulation Effects of FXA Inhibitors (ANNEXA-4) study is an ongoing, multicenter, prospective, open-label, single-group study designed to evaluate the use of AndexXa® in patients with acute major bleeding that was potentially life-threatening.
The authors in this interim report described the outcomes of 67 patients, for whom complete data was available. Patients in this study had acute major bleeding within 18 hours after the administration of one of four Factor Xa inhibitors – ELIQUIS®, XARELTO®, SAVAYSA®, or LOVENOX® (Enoxaparin). Acute major bleeding was defined as potentially life-threatening with signs or symptoms of hemodynamic compromise, decrease in hemoglobin of at least 2 gm/dl, symptomatic bleeding in a critical area or organ. The mean patient age was 77 years, and most patients had a history of cardiovascular disease and thrombotic events and bleeding was predominantly gastrointestinal or intracranial. An independent committee determined hemostatic efficacy on the basis of predetermined criteria. The baseline value for anti-Factor Xa activity was at least 75 ng/ml and 0.5 IU/ml or more for those receiving LOVENOX® and patients had confirmed bleeding severity. All patients received a bolus dose of AndexXa® within 3-6 hours following presentation to the ER followed by a 2-hour infusion of the drug. The two co-primary outcomes were the percent change in the anti-Factor Xa activity and the rate of excellent or good hemostatic efficacy, 12 hours after the AndexXa® infusion. Anti-Factor Xa activity was measured by means of a validated chromogenic assay of Factor Xa enzymatic activity.
It was noted that following the bolus dose of AndexXa®, the median anti-Factor Xa activity decreased by 89% from baseline, among patients receiving XARELTO® and by 93% among patients receiving ELIQUIS® and these levels remained the same during the 2-hour infusion. Four hours after the end of the infusion, the relative decrease in the anti-Factor Xa activity from baseline, among patients receiving XARELTO® was 39% and 30% among those receiving ELIQUIS®. Twelve hours after the AndexXa® infusion, hemostatic efficacy was adjudicated as excellent or good by the independent committee, in 79% of the patients and was consistent across all subgroups. During the 30-day follow up period, thrombotic events occurred in 18% of the patients.
The authors concluded that AndexXa® rapidly reversed anti-Factor Xa activity without significant toxicity, in 79% of the patients, 12 hours after an infusion of AndexXa®. This study also demonstrated that prolonged reversal of Factor Xa inhibition may not be necessary to achieve a good hemostatic response with AndexXa®. Andexanet Alfa for Acute Major Bleeding Associated with Factor Xa Inhibitors. Connolly SJ, Milling TJ, Eikelboom JW, et al. N Engl J Med 2016; 375:1131-1141