SUMMARY: The FDA on May 3, 2018 approved AndexXa® (Andexanet Alfa), a recombinant coagulation Factor Xa, inactivated-zhzo), for patients treated with XARELTO® (Rivaroxaban) and ELIQUIS® (Apixaban), when reversal of anticoagulation is needed due to life-threatening or uncontrolled bleeding. It is estimated that 4 million individuals are presently on Factor Xa inhibitors, and in the US there were approximately 117,000 hospital admissions attributable to Factor Xa inhibitor-related bleeding and nearly 2000 bleeding related deaths per month. There are presently five New Oral Anticoagulants approved in the US for the treatment of Venous ThromboEmbolism (VTE). They include PRADAXA® (Dabigatran), which is a direct thrombin inhibitor and XARELTO® (Rivaroxaban), ELIQUIS® (Apixaban), SAVAYSA® (Endoxaban), BEVYXXA® (Betrixaban) which are Factor Xa inhibitors. Compared to COUMADIN® (Warfarin), the New Oral Anticoagulants have a rapid onset of action, wider therapeutic window, shorter half-lives (7-14 hours in healthy individuals), no laboratory monitoring and fixed dosing schedule. The half-life of these agents can however be prolonged in those with renal insufficiency and may be unsafe and direct oral anticoagulants are ineffective in patients with mechanical heart valves. In several clinical studies, these New Oral Anticoagulants have been shown to reduce the rate of major bleeding by 28% and the rates of intracranial and fatal hemorrhage by 50%, when compared to COUMADIN®. Unlike bleeding caused by COUMADIN®, which can be reversed using Vitamin K or Fresh Frozen Plasma, until now, there were no specific agents available, for reversing bleeding caused by the New Oral Anticoagulants or for stopping the anticoagulant effects of these drugs, in patients who need urgent surgical intervention. The FDA in 2015, granted accelerated approval to PRAXBIND® (Idarucizumab), for the treatment of patients treated with PRADAXA®, a direct thrombin inhibitor, when reversal of the anticoagulant effects of PRADAXA® is needed for emergency surgery/urgent procedures, or in life-threatening or uncontrolled bleeding. However, the Factor Xa inhibitors approved in the US for the treatment of VTE did not have an antidote until this new approval. As such, some Health Care Providers discouraged their patients from taking these direct oral anticoagulants until an antidote became available, should their patients need urgent surgical intervention.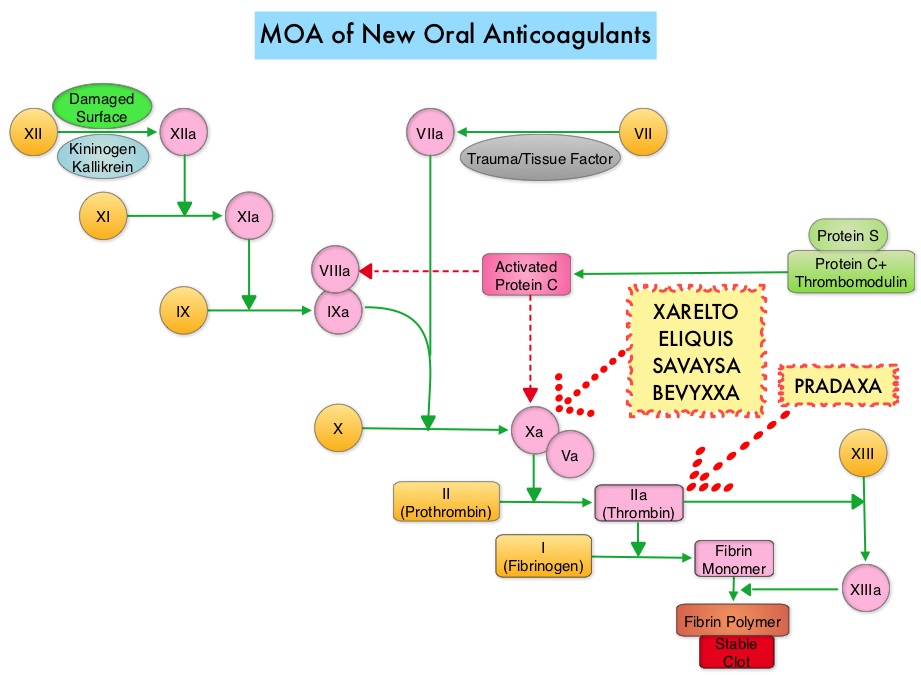
AndexXa® (Andexanet alfa) is a recombinant, modified human Factor Xa decoy protein without intrinsic catalytic activity, that binds Factor Xa inhibitors. The approval of AndexXa® was based on data from two Phase III ANNEXA studies (ANNEXA-A and ANNEXA-R) as well as interim data from the ongoing ANNEXA-4 study. ANNEXA-A and ANNEXA-R are randomized, double-blind, placebo-controlled, Phase III studies which evaluated the safety and efficacy of AndexXa® in reversing the anticoagulant effect of ELIQUIS® and XARELTO® respectively, in healthy volunteers aged 50-68 years. A two-part randomized placebo-controlled study was conducted for each Factor Xa inhibitor, to evaluate AndexXa® administered as a bolus or as a bolus plus a 2-hour infusion. The Primary endpoint was reduction in anti-Factor Xa activity levels, a measure of Factor Xa inhibition by the anticoagulant. Secondary endpoints included reduction in plasma levels of free unbound XARELTO® or ELIQUIS® and restoration of the endogenous thrombin potential (ETP), a measure of thrombin generation.
ANNEXA-A Study: In Part 1, 33 healthy participants were given ELIQUIS® 5 mg twice daily for 3.5 days and then randomized in a 3:1 ratio to receive either AndexXa® administered as a 400 mg IV bolus or placebo. Within 2-5 minutes of completion of the bolus dose, AndexXa® rapidly reduced the anticoagulant activity of ELIQUIS® by 94% compared with placebo (P<0.001), as measured by anti-Factor Xa activity. The reversal of anti-factor Xa activity persisted for 2 hours. Further, AndexXa® significantly reduced the level of free (unbound) ELIQUIS® in the plasma compared with placebo (P<0.001) and fully restored thrombin generation in 100 percent of subjects (P<0.001 vs. placebo). In Part 2, 31 healthy participants received ELIQUIS® 5 mg twice daily for four days and then randomized in a 3:1 ratio to receive either AndexXa® administered as a 400 mg IV bolus followed by a continuous infusion of 4 mg/min for 120 minutes or placebo. AndexXa® significantly reduced anti-Factor Xa activity by 92% compared with placebo (P<0.001), with reversal persisting for 1 to 2 hours after completion of the infusion. The reduction in free unbound ELIQUIS® was sustained with the bolus plus infusion, which significantly reduced the mean plasma concentration of free unbound ELIQUIS® compared with placebo (P<0.001). AndexXa® also restored thrombin generation to normal in all participants who received the compound (p<0.001 vs. placebo).
ANNEXA-R Study: In Part 1, 41 healthy volunteer participants were given XARELTO® 20 mg once daily for four days and then randomized in a 2:1 ratio to receive either AndexXa® administered as an 800 mg IV bolus or placebo. Within 2-5 five minutes of completion of the bolus dose, AndexXa® significantly reversed the anticoagulant activity of XARELTO® by 92% compared with placebo (P<0.001), as measured by anti-Factor Xa activity. Further, AndexXa® significantly reduced the level of free (unbound) XARELTO® in the plasma compared with placebo (P<0.001) and fully restored thrombin generation in 96% of participants (P<0.001 versus placebo). In Part 2, 39 healthy volunteers were given XARELTO® 20 mg once daily for four days and then randomized in a 2:1 ratio to receive either AndexXa® administered as an 800 mg IV bolus followed by a continuous infusion of 8 mg/min for 120 minutes or placebo. AndexXa® significantly reduced anti-Factor Xa activity by 97% compared with placebo (P<0.001), with reversal persisting for 1 to 2 hours after completion of the infusion. The reduction in free unbound XARELTO® was sustained with the bolus plus infusion, which significantly reduced the mean plasma concentration of free unbound XARELTO® compared with placebo (P<0.001). AndexXa® also restored thrombin generation to normal in all participants who received this agent (P<0.001 versus placebo).
ANNEXA-4 Study: This is an ongoing, multicenter, prospective, open-label, single-group study designed to evaluate the use of AndexXa® in patients with acute potentially life-threatening major bleeding, within 18 hours after the administration of one of four Factor Xa inhibitors – ELIQUIS®, XARELTO®, SAVAYSA®, or LOVENOX® (Enoxaparin). . All patients received a bolus dose of AndexXa® within 3-6 hours following presentation to the ER followed by a 2-hour infusion of the drug. The two co-primary outcomes were the percent change in the anti-Factor Xa activity and the rate of excellent or good hemostatic efficacy, 12 hours after the AndexXa® infusion. Anti-Factor Xa activity was measured by means of a validated chromogenic assay of Factor Xa enzymatic activity. Among the 185 evaluable high-risk patients in this open-label study, hemostatic efficacy was adjudicated as excellent or good by the independent committee, 12 hours after the AndexXa® infusion in 83% of patients. It was noted that following the bolus dose of AndexXa®, the median anti-Factor Xa activity decreased by 90% from baseline, among patients receiving XARELTO® and by 93% among patients receiving ELIQUIS® and these levels remained the same during the 2-hour infusion.
In conclusion, AndexXa® is the first and only antidote indicated for patients treated with XARELTO® and ELIQUIS® when reversal of anticoagulation is needed due to life-threatening or uncontrolled bleeding. The availability of this antidote assures both patients and health care providers to consider Factor Xa inhibitors with greater confidence. Andexanet Alfa for the Reversal of Factor Xa Inhibitor Activity. Siegal DM, Curnutte JT, Connolly SJ, et al. N Engl J Med 2015; 373:2413-242. Andexanet Alfa for Acute Major Bleeding Associated with Factor Xa Inhibitors. Connolly SJ, Milling TJ, Eikelboom JW, et al. N Engl J Med 2016; 375:1131-1141