
The FDA on December 14, 2018 approved NPLATE® for pediatric patients 1 year of age and older with Immune Thrombocytopenia (ITP) for at least 6 months, who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. NPLATE® is a product of Amgen Inc.
Author: RR
TECENTRIQ® (Atezolizumab)
The FDA on December 6, 2018 approved TECENTRIQ® in combination with AVASTIN® (Bevacizumab), TAXOL® (Paclitaxel), and Carboplatin for the first-line treatment of patients with metastatic non-squamous, Non-Small Cell Lung Cancer (NSq NSCLC) with no EGFR or ALK genomic tumor aberrations. TECENTRIQ® is a product of Genentech, Inc.
Late Breaking Abstract – ASH 2018 Frontline DARZALEX® with REVLIMID® and Dexamethasone – A New Standard for Transplant-Ineligible Myeloma Patients
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 30,770 new cases will be diagnosed in 2018 and 12,770 patients are expected to die of the disease. Multiple Myeloma (MM) in 2018 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). REVLIMID® (Lenalidomide) based regimens are often prescribed for patients with newly diagnosed, transplant-ineligible Multiple Myeloma. REVLIMID®, a thalidomide analogue has immunomodulatory, tumoricidal, and antiangiogenic properties, and synergizes with Dexamethasone to enhance anti-myeloma activity. DARZALEX® (Daratumumab) is a human IgG1 monoclonal antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Mediated Cytotoxicity and direct apoptosis. Additionally, DARZALEX® may have a role in immunomodulation by depleting CD38-positive regulator Immune suppressor cells, and thereby expanding T cells, in patients responding to therapy.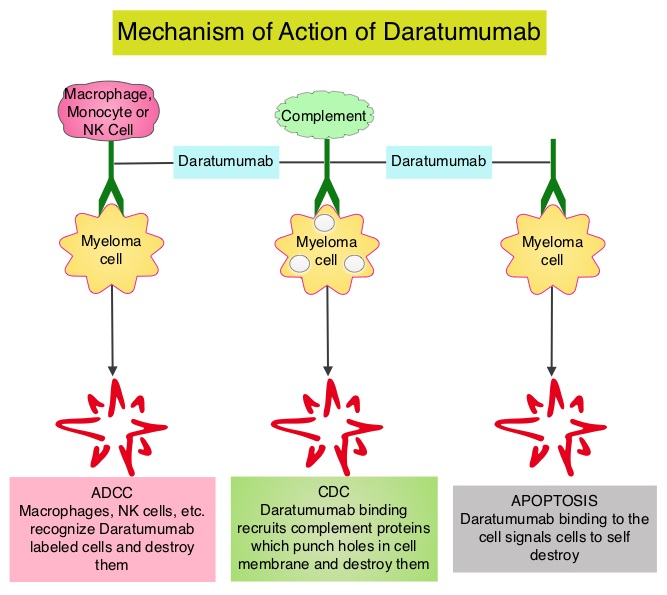
The FDA in May, 2018 approved DARZALEX® in combination with VELCADE® (Bortezomib), a proteasome inhibitor, Melphalan, an alkylating agent and Prednisone (VMP regimen), for the treatment of patients with newly diagnosed Multiple Myeloma who are ineligible for Autologous Stem Cell Transplant (ASCT). VMP regimen however is mostly utilized in Europe and not in the US. In the POLLUX trial, addition of DARZALEX® to REVLIMID® and Dexamathasone (D-Rd) showed the greatest benefit, with a 63% reduction in risk of disease progression or death (HR=0.37; P<0.001) in patients with Multiple Myeloma who had at least one prior line of therapy, compared to REVLIMID® and Dexamathasone (Rd) . Based on the efficacy and tolerable safety profile of D-Rd, the authors conducted a phase III study (MAIA), comparing D-Rd to Rd in transplant-ineligible newly diagnosed Multiple Myeloma patients and reported the prespecified interim analysis of the study.
The MAIA study is a multicenter, international, open-label, phase III trial, which included 737 newly diagnosed Myeloma patients who were not candidates for high-dose chemotherapy and Autologous Stem Cell Transplant (ASCT), due to age 65 years or older or comorbidities. Patients were randomly assigned 1:1 to receive REVLIMID® 25 mg orally on days 1-21 of each 28-day cycle and Dexamethasone 40 mg once a week, with or without DARZALEX®. Patients assigned DARZALEX® (D-Rd regimen) received 16 mg/kg weekly for the first 8 weeks (cycles 1 and 2), every other week for 16 weeks (cycles 3 to 6), and then every 4 weeks (cycle 7 and beyond) until disease progression or unacceptable toxicity. Treatment groups were well balanced. The median patient age was 73 years and only 1% of patients were 65 years of age or less whereas 44% of patients were 75 years or older. Cytogenetic risk level could be determined in 642 patients of the total population. Eighty-six percent (86%) of these patients were standard risk and 14% were considered high risk. The Primary end point was Progression Free Survival (PFS). Key Secondary endpoints included Overall Response Rate (ORR), Minimal Residual Disease (MRD) negativity rate (10-5 sensitivity), and Safety.
The prespecified interim analysis occurred with a median follow up of 28 months. DARZALEX® regimen significantly improved PFS with the median PFS not reached with D-Rd compared with 31.9 months in the Rd group (HR=0.55; P<0.0001). This represented a 45% reduction in the risk of progression or death in patients treated with D-Rd. D-Rd also resulted in deeper responses with a Complete Response (CR) or better rate of 47.6% in the D-Rd group compared with 24.7% in the Rd group (P<0.0001). The Very Good Partial Response (VGPR) or better rate was 79.3% in the D-Rd arm compared with 53.1% in the Rd arm (P<0.0001). The MRD-negative rate was more than threefold higher with D-Rd versus Rd at 24% versus 7%, respectively. Higher rates of neutropenia, and leukopenia were observed in the D-Rd arm and the safety profile was consistent with previously reported DARZALEX® studies.
The authors concluded that the addition of DARZALEX®, to REVLIMID® and Dexamethasone significantly reduced the risk of progression or death by 45% in newly diagnosed Multiple Myeloma patients who are transplant-ineligible and these results support D-Rd as a new standard of care for this patient group. Phase 3 Randomized Study of Daratumumab Plus Lenalidomide and Dexamethasone (D-Rd) Versus Lenalidomide and Dexamethasone (Rd) in Patients with Newly Diagnosed Multiple Myeloma (NDMM) Ineligible for Transplant (MAIA). Facon T, Kumar SK, Plesner T, et al. Presented at: ASH Annual Meeting and Exposition; December 4-8, 2018; San Diego, California. Abstract LBA-2.
Axillary Radiotherapy is an Alternative to Complete Lymph Node Dissection in Early-Stage Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 266,120 new cases of invasive breast cancer will be diagnosed in 2018 and about 40,920 women will die of the disease. Axillary lymph node evaluation is an important part of breast cancer staging and the presence of axillary lymph metastases decreases the 5-year survival rate by 28-40%. Axillary lymph node status remains the most powerful predictor of breast cancer recurrence and survival. Axillary Lymph Node Dissection (ALND) was first advocated in the 18th century as part of the treatment of invasive breast cancer and has been standard practice until 2 decades back. ALND can be associated with significant morbidities such as upper limb lymphedema, pain, and sensitivity disorders and this can have a major psychological impact on breast cancer patients. Sentinel Lymph Node Biopsy (SLNB) which was introduced into clinical practice in the mid 1990’s, however has now become the standard for Stage I and II breast cancer. The sentinel node is the first lymph node(s) to which cancer cells are most likely to metastasize from a primary tumor. With the introduction of intraoperative lymphatic mapping in the 1990s, Sentinel Lymph Node Biopsy (SLNB) has now gained general acceptance and if the sentinel node is negative for metastatic disease or only has minimal disease, then no further axillary surgery is indicated. Unlike Axillary Lymph Node Dissection (ALND), SLNB is associated with a lower incidence of lymphedema/ seroma at the surgery site, paresthesias and restriction of joint movement. Nine randomized clinical trials have not shown any difference in mortality among patients who underwent ALND or SLNB for either lymph node metastases or negative sentinel lymph nodes, validating Sentinel Lymph Node Biopsy (SLNB). The American Society of Clinical Oncology (ASCO) first published guidelines on the use of SLNB for patients with early stage breast cancer in 2005, based on one randomized clinical trial. Since then, ASCO updated Clinical Practice Guideline based on additional information from 9 randomized clinical trials and13 cohort studies pertinent to SLNB and ALND.
Patients with T1-2 tumors with positive Sentinel Lymph Node Biopsy usually undergo complete ALND and there is increasing controversy about whether ALND is always necessary. AMAROS is a multicenter, randomized phase III trial, sponsored by the European Organisation for Research and Treatment of Cancer (EORTC), in which the effectiveness of complete Axillary Lymph Node Dissection (ALND) was compared with axillary Radiation Therapy (RT), in patients with invasive breast cancer. The rationale was that Radiation Therapy uses high-energy x-rays to damage tumor cells and may be a less invasive treatment and causes fewer side effects than complete ALND. This study was conducted to evaluate whether axillary RT could yield comparable outcomes to ALND with fewer adverse side effects, in this patient population. This trial enrolled 4806 patients with early-stage, clinically node-negative breast cancer of whom 1425 patients had a positive sentinel lymph node biopsy. Of these patients, 744 were randomly assigned to undergo complete ALND, whereas 681 patients received axillary RT. Both treatment groups were well balanced. The first 5-year follow up data published in 2013 showed that upper extremity lymphedema occurred significantly less often in those who received Radiotherapy compared with those who underwent complete ALND, and recent Quality of life and morbidity data supported these earlier findings.
The authors herein presented the 10 year follow up data of the AMAROS trial. It was noted that at 10 years, 1.82% of patients assigned to axillary RT had an axillary recurrence compared with 0.93% of those assigned to complete ALND, and this suggested that there was no significant difference (P=0.36). Further, there was no significant difference in the distant metastasis–free survival or Overall Survival between the two treatment groups. The distant metastasis–free survival was 78.2% among those assigned to axillary RT and 81.7% among those assigned to complete ALND and Overall Survival in the two treatment groups was 81.4% and 84.6%, respectively. It was noted that there was a higher 10-year cumulative incidence of second primaries among patients assigned to axillary RT compared with those assigned to complete Axillary Lymph Node Dissection (12% versus 8.3%). It remains unclear whether the addition of the radiation will increase the risk of second primary cancers.
It was concluded that axillary Radiotherapy is noninferior to complete Axillary Lymph Node Dissection in terms of locoregional control and this trial suggests that some patients with a positive sentinel lymph node biopsy may be appropriate candidates for axillary Radiotherapy. Rutgers, E. Radiotherapy or surgery of the axilla after a positive sentinel node in breast cancer patients: 10-year results of the EORTC AMAROS trial. Presented at the 2018 San Antonio Breast Cancer Symposium; December 4-8; San Antonio, Texas.(Abstract GS4-01)
Adjuvant KADCYLA® Superior to HERCEPTIN® in High Risk HER2-Positive Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 266,120 new cases of invasive breast cancer will be diagnosed in 2018 and about 40,920 women will die of the disease. The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. Approximately 15-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. HERCEPTIN® (Trastuzumab) is a humanized monoclonal antibody targeting HER2, and adjuvant and neoadjuvant chemotherapy given along with HERCEPTIN® reduces the risk of disease recurrence and death, among patients with HER2-positive, early stage as well as advanced metastatic breast cancer. Since the approval of HERCEPTIN®, several other HER2-targeted therapies have become available. The duration of adjuvant HERCEPTIN® therapy has been 12 months and this length of treatment was empirically adopted from the pivotal registration trials.
KADCYLA® (Ado-Trastuzumab Emtansine, T-DM1) is an Antibody-Drug Conjugate (ADC) comprised of the antibody HERCEPTIN® and the chemotherapy agent Emtansine, linked together. Upon binding to the HER2 receptor, it not only inhibits the HER2 signaling pathways but also delivers a chemotherapy agent Emtansine, a microtubule inhibitor, directly inside the tumor cells. This agent is internalized by lysosomes and destroys the HER2-positive tumor cells upon intracellular release. In the EMILIA trial, KADCYLA® was associated with significant increase in Overall Survival when compared with TYKERB® (Lapatinib) plus XELODA® (Capecitabine), in HER2-positive metastatic breast cancer patients, who had previously received HERCEPTIN® and a Taxane.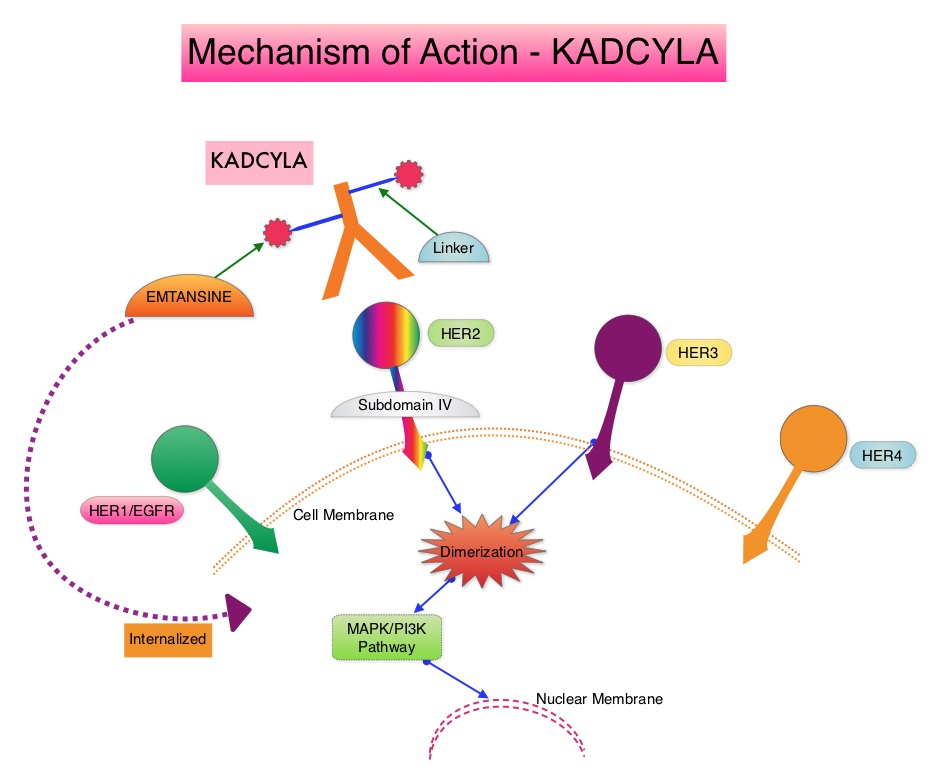
The present study was conducted to address the unmet need of patients who have residual invasive breast cancer after receiving neoadjuvant chemotherapy plus HER2-targeted therapy. These high risk patients have an unfavorable prognosis, compared to those who have no residual cancer following neoadjuvant therapy. The KATHERINE trial is an open-label, phase III global study, which compared KADCYLA® with HERCEPTIN®, as an adjuvant treatment for patients with HER2-positive early breast cancer, who had residual invasive disease following neoadjuvant chemotherapy and HERCEPTIN®.
This study included 1,486 patients with HER2-positive early stage breast cancer, who were found to have residual invasive disease in the breast or axillary lymph nodes at surgery, following at least six cycles (16 weeks) of neoadjuvant chemotherapy with a Taxane (with or without Anthracycline) and HERCEPTIN®. Within 12 weeks of surgery, patients (N=1486) were randomly assigned in a 1:1 ratio to KADCYLA® 3.6 mg/kg IV every 3 weeks or HERCEPTIN® 6 mg/kg IV every 3 weeks, for 14 cycles (743 patients in each group). Both treatment groups were well balanced and hormone receptor positive disease was present in 72% of the patients. The majority of the patients (77%) had received an Anthracycline-containing neoadjuvant chemotherapy regimen, and in 19% of the patients, another HER2-targeted agent in addition to HERCEPTIN® had been administered as a component of neoadjuvant therapy. The Primary end point was invasive Disease Free Survival (defined as freedom from ipsilateral invasive breast tumor recurrence, ipsilateral locoregional invasive breast cancer recurrence, contralateral invasive breast cancer, distant recurrence, or death from any cause). The median duration of follow up was 41.4 months in the KADCYLA® group and 40.9 months in the HERCEPTIN® group.
At the prespecified interim analysis, invasive disease occurred in 12.2% of patients who received KADCYLA® and 22.2% of patients who received HERCEPTIN®. The estimated percentage of patients who were free of invasive disease at 3 years was 88.3% in the KADCYLA® group and 77.0% in the HERCEPTIN® group. Invasive Disease Free Survival which was the Primary end point of the study was significantly higher in the KADCYLA® group than in the HERCEPTIN® group (HR=0.50; P<0.001). This suggested that KADCYLA® reduced the risk of developing an invasive breast cancer recurrence or death by 50%. Distant recurrence as the first invasive disease event occurred in 10.5% of patients in the KADCYLA® group and in 15.9% of the HERCEPTIN® group. A consistent benefit was seen across all prespecified subgroups. Adverse events were consistent with the known safety profile of KADCYLA®, with more toxicities associated with KADCYLA® than with HERCEPTIN®. Additional follow-up will be necessary to determine the Overall Survival benefit with adjuvant KADCYLA®.
It was concluded that among patients with HER2-positive early breast cancer who had residual invasive disease after completion of neoadjuvant therapy, substituting KADCYLA® for adjuvant HERCEPTIN® reduced the risk of recurrence of invasive breast cancer or death was 50%, with the benefit seen across all patient subgroups. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. von Minckwitz G, Huang C-S, Mano MS, et al. for the KATHERINE Investigators. (published online December 5, 2018). New Engl Jour Med. doi: 10.1056/NEJMoa1814017
TECENTRIQ® in Combination with Chemotherapy Improves Overall Survival in Extensive Stage Small-Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Small cell lung cancer (SCLC) accounts for approximately 13-15% of all lung cancers and is aggressive. Patients with extensive stage SCLC are often treated with a combination of Carboplatin or Cisplatin with Etoposide as first line treatment and the tumor response rates are as high as 60-80%. However, majority of the patients relapse within months of completing initial therapy, with a median Overall Survival of approximately 10 months. Based on the premise that SCLC has a high mutation rate, it was hypothesized that these tumors may be immunogenic and could respond to immune-checkpoint inhibitors. This hypothesis was subsequently confirmed in patients with refractory or metastatic SCLC.
TECENTRIQ® (Atezolizumab) is an anti PD-L1 monoclonal antibody that directly binds to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells and blocks the interaction of PD-L1 with PD-1 and B7.1 receptors and thereby enables the activation of T cells and restores tumor-specific T-cell immunity. In a phase I trial, TECENTRIQ® monotherapy demonstrated durable responses, with an acceptable safety profile in patients with relapsed or refractory SCLC. The authors in this study combined checkpoint inhibition with cytotoxic chemotherapy for synergy and improved efficacy. 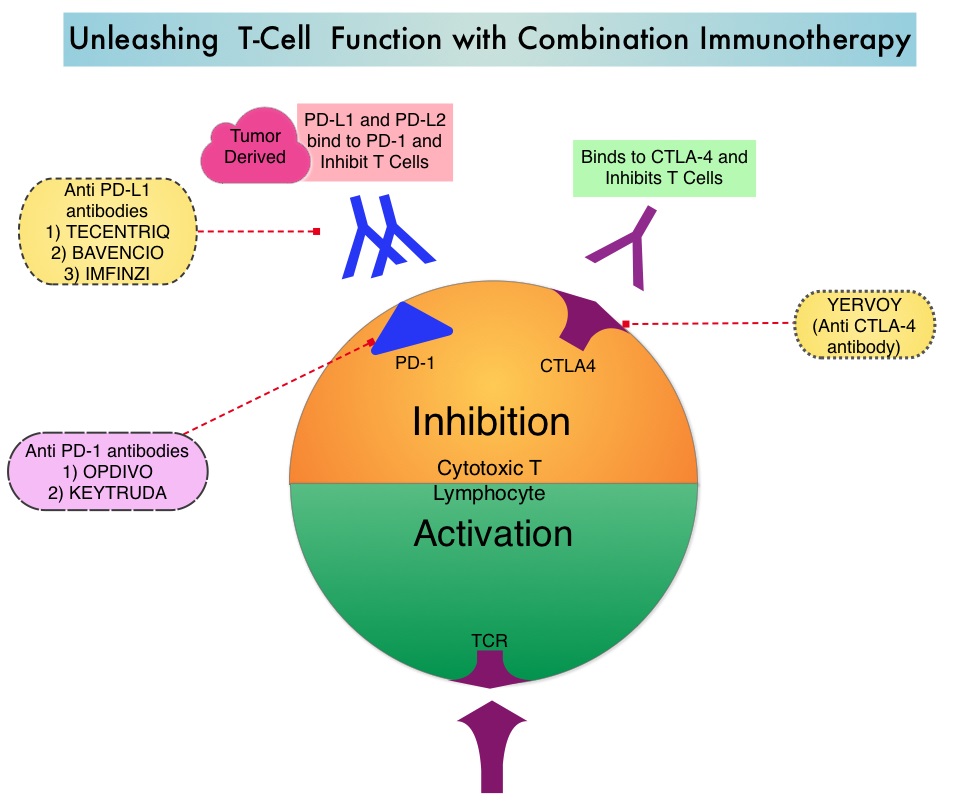
The IMpower133 trial is a multinational, randomized, double-blind, placebo-controlled phase III trial which evaluated the benefit of TECENTRIQ® plus Carboplatin and Etoposide in chemo naïve patients with extensive-stage Small-Cell Lung Cancer. Enrolled patients were randomized in a 1:1 ratio and the induction phase consisted of four cycles of Carboplatin AUC 5 mg/ml/min IV on day 1 and Etoposide 100 mg/m2 IV on days 1-3 of each 21-day cycle, with either TECENTRIQ® 1200 mg IV day 1 of each cycle (N=201) or placebo (N=202). The induction phase was followed by a maintenance phase during which patients received either TECENTRIQ® or placebo (based on previous random assignment) and treatment was continued until disease progression or unacceptable toxicities. The median age was 64 yrs and PD-L1 testing was not a requirement. Prophylactic cranial irradiation was permitted during the maintenance phase of treatment but thoracic radiation therapy was not permitted. The Primary end points were Overall Survival (OS) and Progression Free Survival and Secondary end points included Objective Response Rate (ORR) and Duration of Response.
At a median follow up of 13.9 months, the median OS was 12.3 months in the TECENTRIQ® group compared to 10.3 months in the placebo group (HR=0.70; P=0.007). This suggested a 30% reduction in the risk of death when TECENTRIQ® was added to chemotherapy. The 1-year OS rate was 51.7% in the TECENTRIQ® group and 38.2% in the placebo group. The median Progression Free Survival was also longer in the TECENTRIQ® group than in the placebo group (5.2 months versus 4.3 months, HR=0.77; P=0.02). Survival benefits were consistently seen across patient subgroups. The safety profile of TECENTRIQ® plus Carboplatin and Etoposide was consistent with the previously reported safety profile of the individual agents, with no new findings observed in this trial.
It was concluded that in this multinational trial, the addition of TECENTRIQ® to chemotherapy in the first line treatment of extensive stage Small-Cell Lung Cancer resulted in significantly longer Overall Survival and Progression Free Survival, when compared to chemotherapy alone. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. Horn L, Mansfield AS, SzczÄ™sna A, et al. for the IMpower133 Study Group. N Engl J Med 2018; 379:2220-2229
FDA Approves VITRAKVI®, A Novel Tumor Agnostic Therapy for TRK Fusion-Positive Cancers
SUMMARY: The FDA on November 26, 2018, granted accelerated approval to VITRAKVI® (Larotrectinib) for adult and pediatric patients with solid tumors that have a NeuroTrophic Receptor tyrosine Kinase (NTRK) gene fusion without a known acquired resistance mutation, that are either metastatic or where surgical resection is likely to result in severe morbidity, and who have no satisfactory alternative treatments or whose cancer has progressed following treatment. This is the second tissue-agnostic FDA approval for the treatment of cancer. Tumor genomic profiling enables the identification of specific genomic alterations and thereby can provide personalized treatment options with targeted therapies that are specific for those molecular targets. The FDA in May 2017 granted accelerated approval to KEYTRUDA® (Pembrolizumab), for adult and pediatric patients with unresectable or metastatic, MicroSatellite Instability-High (MSI-H) or MisMatch Repair deficient (dMMR) solid tumors. This was the first FDA approval of a systemic cancer treatment, based on a specific genetic biomarker, independent of tumor origin (first tissue/site-agnostic approval).
A genomic test can be performed on a tumor specimen or on cell-free DNA in plasma (“liquid biopsy”). ImmunoHistoChemistry (IHC) test can be performed on tumor tissue for protein expression that demonstrates a genomic variant known to be a drug target, or to predict sensitivity to a chemotherapeutic drug. Next-generation sequencing (NGS) platforms or second-generation sequencing unlike the first-generation sequencing, known as Sanger sequencing, perform massively parallel sequencing, which allows sequencing of millions of fragments of DNA from a single sample. With this high-throughput sequencing, the entire genome can be sequenced in less than 24 hours. Recently reported genomic profiling studies performed in patients with advanced cancer suggest that actionable mutations are found in 20-40% of patients’ tumors. Next-generation sequencing has enabled the detection of Neurotrophic Tropomyosin Receptor Kinase (NTRK) gene fusions, which was first discovered in colon cancer in 1982. The three TRK family of Tropomyosin Receptor Kinase (TRK) transmembrane proteins TRKA, TRKB, and TRKC are encoded by Neurotrophic Tropomyosin Receptor Kinase genes NTRK1, NTRK2, and NTRK3, respectively. These receptor tyrosine kinases are expressed in human neuronal tissue and are involved in a variety of signaling events such as cell differentiation, cell survival and apoptosis of peripheral and central neurons. They therefore play an essential role in the physiology of development and function of the nervous system. There are over 50 different partner genes that fuse with NTRK genes. Chromosomal fusion involving NTRK genes arise early in cancer development and remain so as tumors grow and metastasize. Gene fusions involving NTRK genes lead to transcription of chimeric TRK proteins which can confer oncogenic potential by increasing cell proliferation and survival. Early clinical evidence suggests that these gene fusions lead to oncogene addiction regardless of tissue of origin. (Oncogene addiction is the dependency of some cancers on one or a few genes for the maintenance of the malignant phenotype). It is estimated that gene fusions involving NTRK genes occurs in about 0.5% to 1% of many common malignancies but in more than 90% of certain rare tumor types, such as salivary gland tumors, a form of juvenile breast cancer, and infantile fibrosarcoma.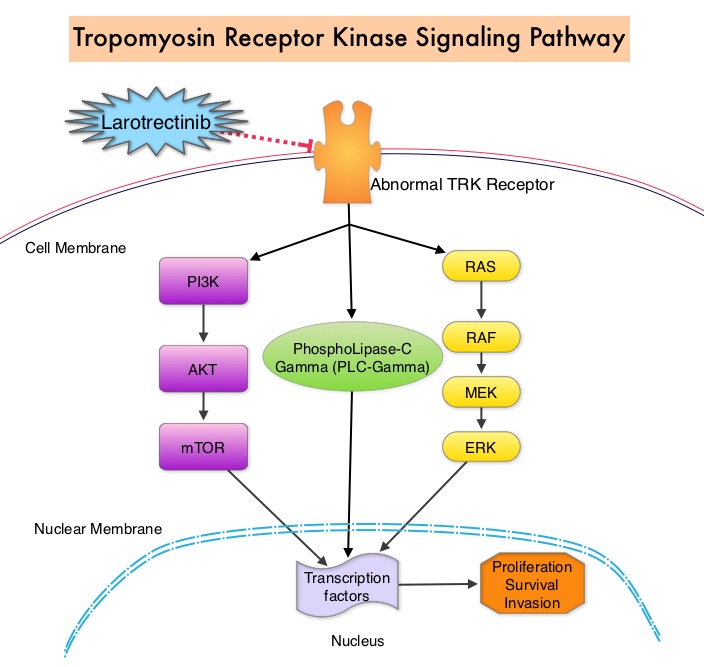
The approval of VITRAKVI®, a potent and highly selective, oral, small molecule inhibitor of all three TRK proteins was based on data from three multicenter, open-label, single-arm clinical trials – LOXO-TRK-14001, a phase I study involving adults, SCOUT, a phase I-II study involving children and NAVIGATE, a phase II study involving adolescents and adults. The authors in this development program included patients of any age and with any tumor type who had chromosomal fusion involving NTRK genes (Age and Tumor agnostic therapy). Positive NTRK gene fusion status was prospectively determined in local laboratories using NGS or Fluorescence In Situ Hybridization (FISH). Treatment efficacy was evaluated in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across the three trials. All patients were required to have progressed following systemic therapy for their disease if available, or would have required surgery with significant morbidity for locally advanced disease. Twelve patients were less than 18 years of age. A total of 12 cancer types were represented, with the most common being salivary gland tumors (22%), soft tissue sarcoma (20%), infantile fibrosarcoma (13%), and thyroid cancer (9%). NTRK gene fusions were inferred in three pediatric patients with infantile fibrosarcoma who had a documented ETV6 translocation by FISH. The Primary end point for the combined analysis was the Overall Response Rate (ORR) according to Independent review. Secondary end points included Duration of Response, Progression Free Survival, and safety.
The ORR was 75%, including 22% Complete Responses and 53% Partial Responses. At the time of database lock, median Duration of Response had not been reached. Response duration was 6 months or longer for 73%, 9 months or longer for 63%, and 12 months or longer for 39% of patients. The safety of VITRAKVI® was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions (20% or more) with VITRAKVI® were fatigue, nausea, vomiting and abnormal liver function studies. None of the patients on VITRAKVI® discontinued therapy, due to a drug-related adverse event.
It was concluded that TRK fusions defined a unique molecular subgroup of advanced solid tumors in children and adults and VITRAKVI® had marked and durable antitumor activity in patients with TRK fusion-positive cancer, regardless of age of the patient or tumor type. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. Drilon A, Laetsch TW, Kummar S, et al. N Engl J Med 2018; 378:731-739
Luspatercept Reduces Blood Transfusion Requirements in MDS and Beta-Thalassemia
SUMMARY: Anemia is a common finding in patients with MyeloDysplastic Syndromes (MDS) and Beta-Thalassemia. These patients are in chronic need for transfusions which in turn can result in iron overload. Erythropoiesis Stimulating Agents (ESAs) are first-line therapy for anemia associated with lower-risk non-del(5q) MDS. ESAs such as Darbepoetin alfa and Epoetin alfa are re-engineered and recombinant DNA technology products of Erythropoietin (EPO), and they stimulate erythropoiesis by binding and activating the EPO receptor. There are however few treatment options for patients who are refractory to, unresponsive to, or ineligible for ESAs. There is therefore an unmet clinical need for safe and effective treatment options, to reduce the RBC transfusion burden in these patients. Beta-Thalassemia is an inherited hemoglobinopathy associated with an erythroid maturation defect and is characterized by ineffective erythropoiesis and impaired RBC maturation.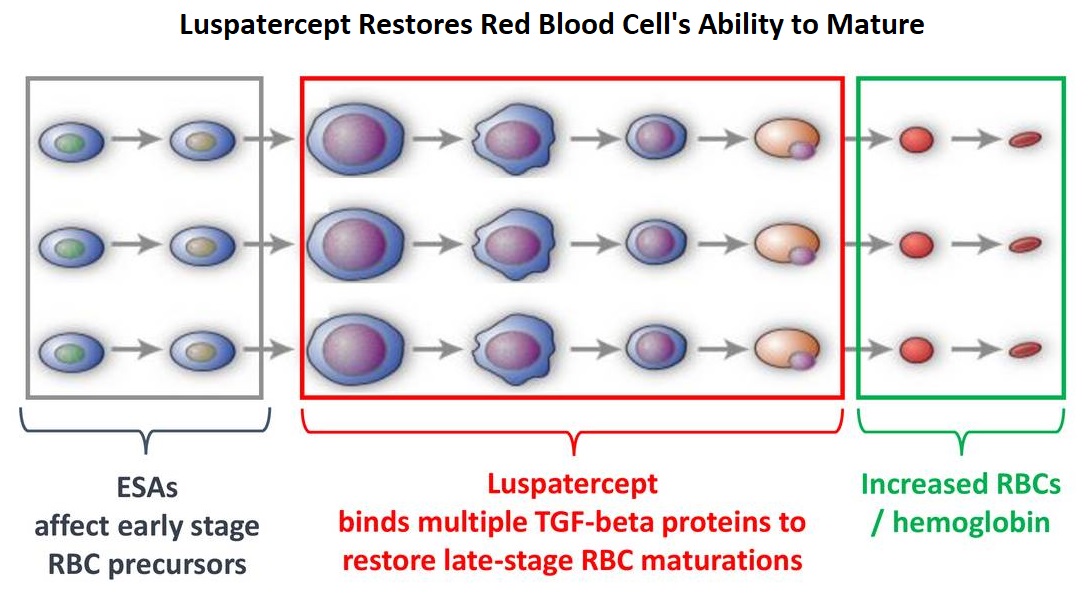
Luspatercept is a soluble fusion protein and is first-in-class erythroid maturation agent that enhances erythropoiesis by promoting late-stage Red Blood Cell precursor differentiation and maturation. It targets select Transforming Growth Factor (TGF)-β superfamily ligands such as GDF11, that regulate late-stage erythropoiesis. This results in a reduction in aberrant Smad2/3 signaling thereby promoting late-stage Red Blood Cell precursor differentiation and maturation. The following two, separate phase III studies have shown reduced blood transfusions requirements in two separate patient populations.
The MEDALIST trial is a randomized, double-blind, placebo-controlled phase III study which evaluated the efficacy and safety of Luspatercept in patients with anemia secondary to MDS, defined as very low-risk, low-risk, or Intermediate-risk with Ring Sideroblasts, according to the Revised International Prognostic Scoring System. Eligible patients were refractory, intolerant, or ineligible to receive ESAs and required RBC transfusions. A total of 229 patients (N=229) were randomized 2:1 to receive either Luspatercept at a starting dose level of 1mg/kg SC with titration up to 1.75 mg/kg if needed (N=153), or placebo SC (N=76), every 3 weeks for 24 weeks or more. The median age was 71 years and median time from diagnosis was 41.8 months. Approximately 95% of patients had previously received ESAs and 90% had an SF3B1 mutation. The Primary endpoint was RBC transfusion independence for 8 weeks or more between week 1 and 24. A key secondary endpoint was RBC transfusion independence for 12 weeks or more between week 1 and 24.
Among those receiving Luspatercept, 38% achieved the Primary endpoint of RBC transfusion independence for 8 weeks or more compared with 13.2% receiving placebo (P<0.0001). Further among those receiving Luspatercept, 28.1% achieved the key secondary endpoint of RBC transfusion independence for 12 weeks or more compared with 7.9% receiving placebo (P=0.0002). Additionally, patients receiving Luspatercept were more likely to achieve an mHI-E (modified hematologic improvement-erythroid) response, defined as a reduction in transfusion of 4 or more RBC units per 8 weeks or a mean hemoglobin increase of 1.5 g/dL or more per 8 weeks in the absence of transfusions, compared with patients receiving placebo (52.9% versus 11.8% during weeks 1-24; P<0.0001).
It was concluded that treatment with Luspatercept significantly decreased transfusion requirements among patients with low or Intermediate-risk MDS with Ring Sideroblasts.
The BELIEVE trial is a randomized, double-blind, placebo-controlled phase III study conducted to determine the efficacy and safety of Luspatercept in adult Beta-Thalassemia patients requiring regular RBC transfusions. In this study, 336 patients with Beta-Thalassemia or Hemoglobin E/ Beta-Thalassemia were randomized in a 2:1 to receive Luspatercept, at a starting dose of 1mg/kg with titration up to 1.25 mg/kg, or placebo, SC every 3 weeks for 48 weeks or more. Patients in both treatment groups continued to receive RBC transfusions and iron chelation therapy to maintain the same baseline Hgb level. Enrolled patients were 18 years or older and required regular RBC transfusions of 6-20 units in the 24 weeks prior to randomization with no transfusion-free period 35 days or more during that time. The median age was 30 years and 58% of patients were female. Patients received a median of 6 RBC units in the 12 weeks prior to treatment and 58% of patients in each treatment group had undergone splenectomy. The Primary endpoint was a 33% or more reduction in transfusion burden (with a reduction of 2 or more RBC units) during weeks 13–24, when compared with a 12-week baseline period.
It was noted that 21.4% of patients in the Luspatercept group achieved the Primary endpoint compared with 4.5% patients in the placebo group (P<0.0001). Towards the end of the trial, 20% of patients overall had decreased their transfusion units by one third or more, and 10% of patients had decreased their transfusions units by half or more. Overall, 70.5% of patients receiving Luspatercept achieved a 33% or more RBC transfusion reduction over any consecutive 12 weeks compared with 29.5% patients receiving placebo (P<0.0001).
It was concluded that treatment with Luspatercept resulted in significant reductions in RBC transfusion requirement, in adults with transfusion-dependent Beta-Thalassemia.
The most common adverse events included fatigue and muscle pain. It remains to be seen if Luspatercept would have similar efficacy in patients with high-risk MDS and patients with lower-risk MDS without ring sideroblasts.
The Medalist Trial: results of a phase 3, randomized, double-blind, placebo-controlled study of luspatercept to treat anemia in patients with very low-, low-, or intermediate-risk myelodysplastic syndromes (MDS) with ring sideroblasts (RS) who require red blood cell (RBC) transfusion. Fenaux P, Platzbecker U, Mufti GJ, et al. Presented at: 2018 ASH Annual Meeting; Dec. 1-4, 2018; San Diego. Abstract 1. https://ash.confex.com/ash/2018/webprogram/Paper110805.html
The Believe Trial: Results of a Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of Luspatercept in Adult Beta-Thalassemia Patients Who Require Regular Red Blood Cell (RBC) Transfusions. Cappellini MD, Viprakasit V, Taher A, et al. Presented at: 2018 ASH Annual Meeting; Dec. 1-4, 2018; San Diego. Abstract 163. https://ash.confex.com/ash/2018/webprogram/Paper112435.html
FDA Approves VENCLEXTA® for Elderly Patients with AML
SUMMARY: The FDA on November 21, 2018 granted accelerated approval to VENCLEXTA® (Venetoclax) in combination with Azacitidine or Decitabine or low-dose Cytarabine for the treatment of newly diagnosed Acute Myeloid Leukemia (AML) in adults who are age 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy. The American Cancer Society estimates that in 2018, 19,520 new cases of Acute Myeloid Leukemia (AML) will be diagnosed in the United States and 10,670 patients will die of the disease. AML can be considered as a group of heterogeneous diseases with different clinical behavior and outcomes. A significant percentage of patients with newly diagnosed AML are not candidates for intensive chemotherapy. Even with the best available therapies, the 5 year Overall Survival in patients 65 years of age or older is less than 5%.
The pro-survival (anti-apoptotic) protein BCL2 is over expressed by AML cells and regulates clonal selection and cell survival. A new class of anticancer agents known as BH3-mimetic drugs mimic the activity of the physiologic antagonists of BCL2 and related proteins and promote apoptosis (programmed cell death). VENCLEXTA® is a second generation, oral, selective, small molecule inhibitor of BCL2 and restores the apoptotic processes in tumor cells.
The present FDA approval was based on two open-label non-randomized clinical trials in patients with newly diagnosed AML who were 75 years of age or older or had comorbidities that precluded the use of intensive induction chemotherapy. Efficacy was established based on the rate of Complete Remission (CR) and CR duration.
The M14-358 study is an open-label, phase Ib dose escalation and expansion study which evaluated the safety and efficacy of VENCLEXTA® in combination with HypoMethylating Agents, Azacitidine or Decitabine. This study included a subpopulation of 80 patients who received VENCLEXTA® (daily ramp-up to a final dose of 400 mg once daily) in combination with a hypomethylating agent, either Azacitidine (N=67) or Decitabine (N=13). Patients were hospitalized for monitoring during the ramp-up and received prophylaxis for Tumor Lysis Syndrome. Azacitidine was administered at 75 mg/m2 SC or IV on days 1-7 of each 28-day cycle and Decitabine was administered at 20 mg/m2 IV on days 1-5 of each 28-day cycle. Treatment was continued until disease progression or unacceptable toxicity. Majority of patients in each treatment group had an ECOG performance status of 0 or 1. In the Azacitidine group, 64% of patients had intermediate cytogenetic risk and 34% had poor cytogenetic risk whereas this was 38% and 62%, respectively in the Decitabine group.
The median follow up was 7.9 months for the Azacitidine group and 11 months for the Decitabine group. The Complete Response rate was 37% in the Azacitidine group with a median observed time in remission of 5.5 months. The rates of CR with partial hematologic recovery were 24%. In combination with Decitabine, the CR rate was 54%, with a median observed time in remission of 4.7 months. The CR with partial hematologic recovery was 7.7%.
The M14-387 study is an open-label, phase Ib/II dose escalation and expansion study which evaluated the safety and efficacy of VENCLEXTA® in combination with Low Dose Ara-C (Cytarabine). This study included a subpopulation of 61 AML patients who received VENCLEXTA® plus low-dose Cytarabine. Included patients had newly diagnosed AML, and some patients had previous exposure to a HypoMethylating Agent for an antecedent hematologic disorder. Patients received VENCLEXTA® (daily ramp-up to a final dose of 600 mg orally once daily). Patients were hospitalized for monitoring during the ramp-up and received prophylaxis for Tumor Lysis Syndrome. Cytarabine was given at 20 mg/m2 SC on days 1-10 of each 28-day cycle. Treatment was continued until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21%, with a median observed time in remission of 6 months. The rate of CR with partial hematologic recovery was 21%. The most common adverse reactions to VENCLEXTA® in combination with Azacitidine, Decitabine or low-dose Cytarabine were fever, nausea, vomiting, diarrhea, fatigue, cytopenias, myalgias, dyspnea, peripheral edema and hypotension.
This FDA approval marks a significant advance for patients with Acute Myeloid Leukemia, who are unable to tolerate standard intensive induction chemotherapy. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm626499.htm
The International Association for the Study of Lung Cancer Issues Statement on Lung Cancer Screening CALL TO ACTION
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer related mortality in the United States. Lung cancer is a growing global epidemic with 1.6 million deaths annually. Over 60% of individuals present with advanced disease at the time of diagnosis and this can result in poor outcomes. Early detection can however lead to lowered mortality. Implementing a validated tool to reliably detect early stage, curable lung cancer has been a priority of the International Association for the Study of Lung Cancer (IASLC), in its mission to conquer thoracic cancers worldwide.
The IASLC on October 25, 2018 issued a statement on lung cancer screening with Low-Dose Computed Tomography (LDCT), based on results from the Dutch-Belgian NELSON lung cancer screening trial presented at the IASLC 19th World Conference on Lung Cancer (WCLC) in Toronto, Canada. The IASLC is the only global organization dedicated solely to the study of lung cancer and other thoracic malignancies and includes more than 7,500 lung cancer specialists across all disciplines in over 100 countries.
EVIDENCE:
The National Lung Cancer Screening Trial (NLST) demonstrated that annual lung cancer screening with Low-Dose CT (LDCT) reduced lung cancer mortality by 20% and overall mortality by 7% compared with controls. Based on the NLST results, NCCN issued guidelines recommending LDCT in 2011, USPSTF (United States Preventive Services Task Force) recommended lung cancer screening with LDCT in high risk patients in 2013 and Low-Dose CT screening was approved in the United States for those at high risk (between the ages of 55 and 77 and a smoking history of 30 pack-years or more and not have quit within the past 15 years).
The Dutch-Belgian Lung Cancer Screening Trial (NELSON) is Europe’s largest lung cancer screening trial and enrolled 15,792 individuals at high risk for lung cancer. Data from this study was presented at the World Conference on Lung Cancer this year which decisively confirmed that annual lung cancer screening with Low-Dose CT in high-risk patients ((age 50-74 years, more than 10 cigarettes/day for more than 30 years or more than 15 cigarettes/day for more than 25 years), reduced lung cancer deaths by 26% in men and up to 61% in women.
RECOMMENDATIONS:
With two trials from the United States and Europe demonstrating significant mortality reduction in high risk, tobacco-exposed populations, IASLC emphasizes that early detection must be routinely provided along with best-practice smoking cessation, to enable optimal health outcomes in the setting of individuals who continue to consume tobacco products. Acknowledging that for implementation of Low-Dose CT screening worldwide, each national health service has the authority to decide its own course of action, IASLC has urged its members and others around the world to implement screening programs that incorporate a multidisciplinary group of experts and use best practice in screening care, with focus on the following:
• Identification of high-risk individuals
• Acquisition of consistent high-quality images (from Low-Dose CT) and incorporation of radiologic guidelines, including definitions for positive versus negative results
• Use of defined clinical workup for indeterminate nodules and for pathology reporting of nodules
• Incorporation of a defined process for surgical or other diagnostic interventions of suspicious nodules
• Integration of smoking cessation into lung cancer CT screening programs
It was concluded that based on the data from these two large, well designed US and European randomized trials, the WCLC committee’s screening experts came to an unanimous consensus that now is the time for international leaders, governments, health care systems and other stakeholders to implement global lung cancer screening programs, as they do for breast cancer (mammography) and colon cancer (colonoscopy), which save the thousands of lives. https://www.iaslc.org/news/iaslc-issues-statement-lung-cancer-screening-low-dose-computed-tomography