SUMMARY: The American Cancer Society estimates that for 2018, about 20,940 new cases of Chronic Lymphocytic Leukemia (CLL) will be diagnosed in the US and 4,510 patients will die of the disease. CLL accounts for about 25% of the new cases of leukemia and the average age at the time of diagnosis is around 71 years. B-cell CLL is the most common type of leukemia in adults, accounting for about 11% of all hematologic malignancies. Chemoimmunotherapy with Fludarabine, Cyclophosphamide, and Rituximab (FCR) has long been the gold standard and the most commonly used treatment regimen for younger, fit, treatment naïve patients with chronic lymphocytic leukemia. This is based on phase III trial data showing improvement in both Progression Free Survival (PFS) and Overall Survival (OS) compared with chemotherapy alone. FCR regimen however is associated with significant toxicities and cannot be tolerated by all CLL patients. IMBRUVICA® (Ibrutinib) is an oral, irreversible inhibitor of BTK and inhibits cell proliferation and promotes programmed cell death (Apoptosis) by blocking B-cell activation and signaling. IMBRUVICA® in phase III trials showed improved PFS and OS when compared to Chlorambucil in previously untreated, elderly patients with CLL. Nonetheless, the efficacy of IMBRUVICA® as a first-line treatment for younger CLL patients (70 years or younger), compared to the most efficacious regimen such as FCR, is unknown.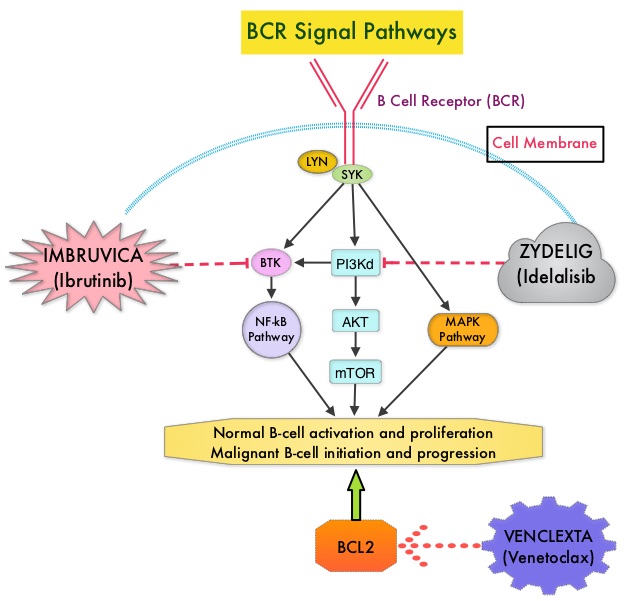
E1912, led by the ECOG-ACRIN Research Group (ECOG-ACRIN), is a randomized phase III study in which IMBRUVICA® (Ibrutinib) plus RITUXAN® (Rituximab) was compared to Fludarabine, Cyclophosphamide, and RITUXAN® (FCR) chemotherapy regimen, in previously untreated patients aged 70 years or younger with Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL). In this trial, 529 patients were randomly assigned in a 2:1 ratio to receive IMBRUVICA® 420 mg orally daily until disease progression along with RITUXAN® 50 mg/m2 on day 1 of cycle 2, 325 mg/m2 on day 2 of cycle 2, 500 mg/m2 on day 1 of cycles 3-7 (N=354) or six courses of Fludarabine 25 mg/m2 IV along with Cyclophosphamide 250 mg/m2 IV days 1-3 and RITUXAN® 50 mg/m2 IV on day 1 of cycle 1, 325 mg/m2 on day 2 of cycle 1, 500 mg/m2 on day 1 of cycles 2-6, given every 28 days (N=175). The median age was 58 years and 40% of the patients were 60 years of age or older. The Primary endpoint was Progression Free Survival (PFS) and the Secondary endpoint was Overall Survival (OS).
With a median follow up of 33.4 months, at the first interim analysis, IMBRUVICA® plus RITUXAN® significantly improved PFS compared to FCR (HR=0.35; P<0.0001), with a 65% reduction in the risk of progression or death with IMBRUVICA® plus RITUXAN® compared with FCR. The combination of IMBRUVICA® plus RITUXAN® also demonstrated improved OS (HR=0.17; P=0.0003) and this suggested that IMBRUVICA® plus RITUXAN® combination reduced the risk of death by 83% compared with FCR. In a subgroup analysis, the PFS benefit with IMBRUVICA® plus RITUXAN® was seen independent of age, sex, Performance Status (0-2), disease stage, as well as presence or absence of cytogenetic abnormality, deletion 11q23. At the time of this analysis, IMBRUVICA® plus RITUXAN® was also superior to FCR among IGHV unmutated patients (HR=0.26; P<0.0001), suggesting a 74% reduction in the risk of progression or death with IMBRUVICA® plus RITUXAN®, compared to FCR. A statistically significant benefit however was not observed among those with IGHV mutations, although there was a positive trend noted (HR=0.44; P=0.07). Treatment-related Grade 3 and 4 toxicities were significantly lower with IMBRUVICA® compared with FCR (58% versus 72%, respectively; P=0.004). FCR was more frequently associated with Grade 3 and 4 neutropenia (44% versus 23%) as well as infectious complications (18% versus 7%).
It was concluded that a combination of IMBRUVICA® and RITUXAN®, significantly improved PFS and OS, when compared to FCR, with fewer toxicities, among patients 70 years of age or under, with previously untreated CLL. The authors noted that these findings have immediate practice changing implications and establish IMBRUVICA® – based therapy as the most effective first-line therapy for untreated patients with CLL. Randomized Phase III Study of Ibrutinib (PCI-32765)-Based Therapy Vs. Standard Fludarabine, Cyclophosphamide, and Rituximab (FCR) Chemoimmunotherapy in Untreated Younger Patients with Chronic Lymphocytic Leukemia (CLL): A Trial of the ECOG-ACRIN Cancer Research Group (E1912. Shanafelt TD, Wang V, Kay NE, et al. Presented at the 2018 ASH Annual Meeting. December 1-4, 2018; San Diego. Abstract LBA-4.