
SUMMARY: Hemophilia A is an X-linked, recessive bleeding disorder caused by mutations in the gene encoding coagulation Factor VIII, resulting in deficiency of functional plasma clotting Factor VIII (FVIII). Hemophilia A can also arise from spontaneous mutation in the gene encoding coagulation Factor VIII. Patients with severe Hemophilia A (Factor VIII activity level less than 1 IU/dl) are at a high risk for spontaneous bleeding in joints and soft tissue, resulting in painful disabling arthropathy as well as increased risk of intracranial hemorrhage and early death. These patients often are on prophylactic Factor VIII therapy and may also require on-demand therapy with Factor VIII for breakthrough bleeding. In spite of the availability of Factor VIII concentrates with extended half-life, patients still require frequent infusions, to maintain adequate hemostasis. This can have a negative impact on quality of life and additionally can result in the development the alloantibodies (inhibitors) that can neutralize the effect of exogenously administered replacement clotting factors.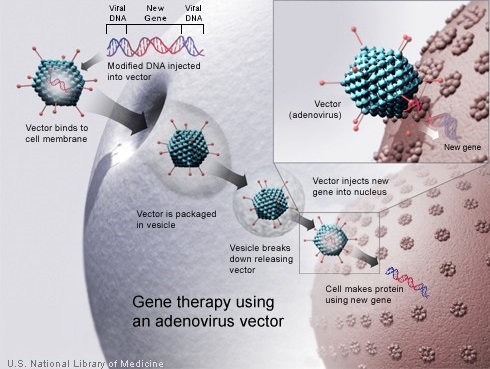
Gene therapy enables the introduction of a normal copy of the gene into the cells and thereby restores the function of the abnormal or missing protein. Because a gene cannot be inserted directly into a cell, a carrier called a vector is genetically engineered to deliver the gene. Majority of gene therapy clinical trials employ viruses as vectors, as these viruses are able to deliver the new gene by infecting the cell and hijacking the cellular machinery for propagation. The viruses are modified to prevent them from causing disease. As vectors, the most extensively studied viruses are Retroviruses, Adenoviruses and Adeno-Associated Viruses (AAVs). Retroviruses integrate their genetic material (including the new gene) into a chromosome in the human cell, whereas Adenoviruses introduce their DNA into the nucleus of the cell, but the DNA is not integrated into a chromosome. The vector can be injected directly into a specific tissue in the body or given intravenously. Alternately, a sample of the patient’s cells can be exposed to the vector in the lab and the cells containing the vector can then be given back to the patient. If the treatment is successful, the new gene delivered by the vector, will start making a functioning protein.
The authors in this study developed AAV5-hFVIII-SQ (valoctocogene roxaparvovec), an AAV serotype 5 vector, in order to overcome specific challenges such as the large size of the Factor VIII coding region and inefficient expression of the human Factor VIII coding sequence. They then conducted a dose-escalation study involving nine patients. Eligible patients were adults with severe Hemophilia A, with no history of Factor VIII inhibitor development and without detectable immunity to the AAV5 capsid. Enrolled patients received a single IV infusion of AAV5-hFVIII-SQ and participants were enrolled sequentially into Low dose, Intermediate dose and High dose cohorts, and the follow up period was 52 weeks. All patients who had been receiving prophylactic Factor VIII therapy previously were withdrawn from prophylaxis. Patients were however permitted to self-administer Factor VIII therapy in the event of bleeding after gene transfer. The Primary end point was safety and the Primary efficacy goal was a Factor VIII activity level of at least 5 IU/dl, at week 16 after gene transfer. Secondary efficacy measures included the frequency of Factor VIII use and the number of bleeding episodes.
It was noted that in the High dose cohort (6×1013 vector genomes-vg/kg), the Factor VIII activity level was more than 5 IU/dl between weeks 2 and 9 after gene transfer and the level in six patients normalized (more than 50 IU/dl) and was maintained at 1 year after treatment with a single dose. Further in this high dose cohort, the median annualized bleeding rate among patients who had previously received prophylactic therapy decreased from 16 events before the study to 1 event after gene transfer, and by week 22, none of the patients in this cohort reported the use of Factor VIII for bleeding. The main adverse event was a slight elevation in the serum alanine aminotransferase (ALT) level. Neutralizing antibodies (inhibitors) to Factor VIII were not detected.
The authors concluded that gene therapy with a high dose infusion of AAV5-hFVIII-SQ was associated with the normalization of Factor VIII activity level over a period of 1 year with stabilization of hemostasis and a profound reduction in Factor VIII use. This landmark study has the potential to pave the way for a cure for Hemophilia patients. AAV5–Factor VIII Gene Transfer in Severe Hemophilia A. Rangarajan S, Walsh L, Lester W, et al. N Engl J Med 2017; 377:2519-2530
