CheckMate-9ER primary and exploratory findings: baseline characteristics and treatment outcomes, including depth and durability of response with cabozantinib plus nivolumab
Written by Dr Manojkumar Bupathi
Sponsored by Exelixis, Inc.
Baseline disease characteristics and tumor biology play a pivotal role in shaping outcomes for patients with advanced renal cell carcinoma (aRCC). As treatment paradigms have evolved toward combination strategies, it has become increasingly important not only to demonstrate efficacy in the overall population, but also to consider the potential for the depth, durability, and consistency of responses across clinically relevant subgroups.1,2
The Phase 3 CheckMate-9ER trial established cabozantinib plus nivolumab as a first-line option for patients with aRCC, demonstrating significant improvements in progression-free survival, overall survival, and objective response rate compared with sunitinib. However, beyond these pivotal results, additional insights from descriptive analyses on long-term outcomes and response characteristics are noteworthy to help inform real-world clinical decision-making.3
In this article, we review the key findings from the CheckMate-9ER trial, including updated 5-year follow-up data, and explore post hoc analyses evaluating baseline characteristics associated with response. In particular, we focus on patients achieving complete responses and examine the depth and durability of response to better contextualize the long-term clinical data observed with cabozantinib plus nivolumab.1,4,5
Overview of CheckMate‐9ER
Cabozantinib is a tyrosine kinase inhibitor, targeting MET, AXL, and VEGFR. Based on the results from the CheckMate-9ER trial, a Phase 3, randomized, open-label, head-to-head trial versus sunitinib, cabozantinib + nivolumab was approved for patients with previously untreated aRCC.3,4
We will begin by reviewing the results from the pivotal trial CheckMate-9ER, which enrolled 651 patients with previously untreated aRCC containing a clear-cell component. Participants were randomized 1:1 to receive cabozantinib 40 mg once daily plus nivolumab 240 mg every 2 weeks, or sunitinib 50 mg once daily administered on a 4 weeks on, 2 weeks off schedule.3,4
- Endpoints assessed3:
- Primary endpoint: PFS*
- Secondary endpoints: OS, ORR*, and safety
*Assessed by BICR.4
CheckMate-9ER studied a broad patient population, including those with a high burden of disease. Select baseline characteristics are shown in Table 1 for the cabozantinib + nivolumab arm.3
Table 13
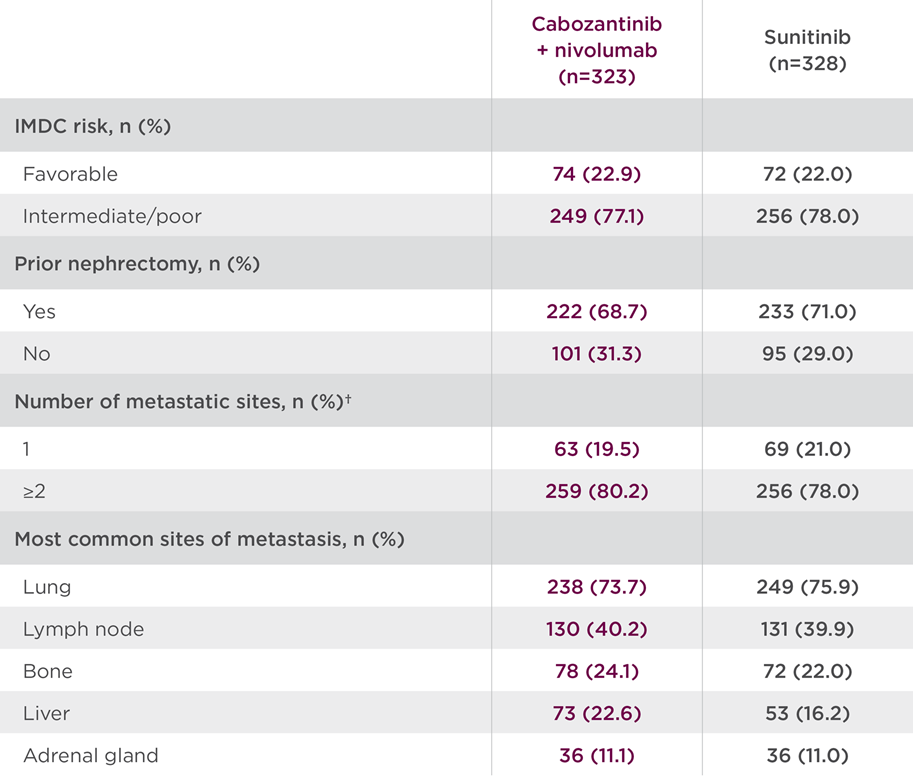
†Data are for tumor sites defined at baseline by the investigators according to RECIST v1.1. The number of target or nontarget lesions at baseline was not reported for 1 patient in the CABOMETYX + OPDIVO group and for 3 patients in the sunitinib group.3
Results in the CheckMate-9ER primary analysis of the intent-to-treat population (median follow-up of 18.1 months; range: 10.6-30.6 months) were as follows3:
- Median PFS* (mPFS) was 16.6 months with cabozantinib + nivolumab (95% CI: 12.5-24.9) vs 8.3 months with sunitinib (95% CI: 7.0-9.7) (HR=0.51; 95% CI: 0.41-0.64; P<0.0001)4,6
- Median OS (mOS) was not reached in either arm (HR=0.60; 98.89% CI: 0.40-0.89; P=0.001)4,6
- Pre-planned final analysis (median follow-up: 32.9 months; range: 25.4-45.4 months): mOS was 37.7 months for the combination (95% Cl: 35.5-NR) vs 34.3 months with sunitinib (95% Cl: 29.0-NR) (HR=0.70; 95% CI: 0.55-0.90)
- ORR* was 55.7% (95% CI: 50.1-61.2) with the combination vs 27.1% with sunitinib (95% CI: 22.4-32.3; P<0.0001)4,6
- Complete response (CR) was 8% (n=26/323) for the combination arm vs 4.6% (n=15/328) with sunitinib
- Partial response (PR) was 48% (n=154/323) for the combination arm vs
23% (n=74/328) with sunitinib
- Serious adverse reactions occurred in 48% of patients receiving cabozantinib + nivolumab (n=320). The most frequent serious adverse reactions reported in ≥2% of patients were diarrhea, pneumonia, pneumonitis, pulmonary embolism, urinary tract infection, and hyponatremia. Fatal intestinal perforations occurred in 3 (0.9%) patients4
- The most common adverse reactions (≥20%) were diarrhea (64%), fatigue (51%), hepatotoxicity (44%), palmar‐plantar erythrodysesthesia (PPE) (40%), stomatitis (37%), rash (36%), hypertension (36%), hypothyroidism (34%), musculoskeletal pain (33%), decreased appetite (28%), nausea (27%), dysgeusia (24%), abdominal pain (22%), upper respiratory tract infection (20%), and cough (20%)4
The results of the CheckMate-9ER 5-year follow-up analysis (median follow-up of 67.6 months; range 60.2-80.2 months) were as follows. No formal statistical testing was conducted at the time of the updated analysis5:
- mPFS* was 16.4 months with the combination (95% CI: 12.5-19.3) vs 8.3 months with sunitinib (95% CI: 7.0-9.7) (HR=0.58; 95% CI: 0.49-0.70)
- mOS was 46.5 months with the combination (95% CI: 40.6-53.8) vs 35.5 months with sunitinib (95% CI: 29.2-42.8) (HR=0.79; 95% CI: 0.65-0.96)
- ORR* was 55.7% (95% CI: 50.1-61.2) for the combination arm vs 27.4% with sunitinib (95% CI: 22.7-32.6)
- CR was 13.9% (n=45/323) for the combination arm vs 4.6% (n=15/328)
with sunitinib
- PR was 41.8% (n=135/323) for the combination arm vs 22.9% (n=75/328)
with sunitinib
- Safety and tolerability were similar to previous follow-ups
*Assessed by BICR.4
To further explore long-term data from the CheckMate-9ER trial, post hoc analyses were conducted to evaluate the baseline characteristics of patients who achieved CR, assess depth of response (DepOR) among those patients with PR, and examine the duration of response in patients with CR or PR.1
Depth and durability of response in patients treated with cabozantinib + nivolumab
The exploratory, post hoc analyses of the 5-year CheckMate-9ER data evaluated depth and durability of response in patients randomized to cabozantinib + nivolumab who were alive 6 months after randomization and who had available data for best overall response (BOR) and sum of diameters of target lesions.1
- Of the 323 patients randomized to cabozantinib + nivolumab, 293 were alive at the
6-month landmark
- Patients were categorized into DepOR subgroups based on BOR per BICR using the maximum tumor reduction observed at any time during the study period
Baseline demographic and disease characteristics of patients receiving cabozantinib + nivolumab by DepOR subgroup are shown in Table 2, which includes patients with characteristics associated with poor prognosis.1
Table 21,7
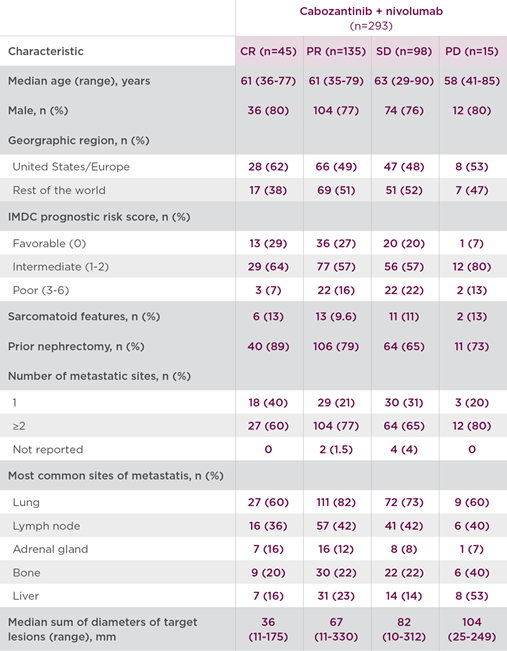
- Among patients who achieved CR (n=45), the median duration of response was not reached at the time of analysis (95% CI: 30.5-NE)1*
- Among patients who achieved PR (n=135), the median duration of response was 17.3 months (95% CI: 13.8-20.7)7*
- Median duration of treatment with cabozantinib + nivolumab was 30.4 months for patients achieving CR, over 2 years for those with PR, and 15.3 months for those with stable disease (SD)1*
- Safety was similar across all subgroups achieving an objective response, and the most frequently reported Grade 3/4 treatment-related adverse events included hypertension (13%), diarrhea (9%), hyponatremia (8%), and PPE (8%)1
These post hoc exploratory analyses are descriptive in nature. No statistical procedure was employed. Results should be considered hypothesis generating.
*Patients who were alive at the 6-month timepoint.1,7
Discussion
In the primary analysis cabozantinib + nivolumab doubled mPFS and demonstrated superior efficacy vs sunitinib across OS and ORR.4 Building on these results, the primary results and exploratory analyses from CheckMate-9ER provide additional context to the established efficacy of cabozantinib + nivolumab in the first-line treatment of aRCC.1,4 Notably, meaningful responses were observed among patients with baseline features traditionally associated with poorer prognosis, underscoring the broad applicability of this combination.1
The median duration of response was not reached among patients achieving complete response. The durability of responses and extended treatment exposure did not appear to be associated with a disproportionate increase in toxicity.1
These findings are derived from post hoc, descriptive exploratory analyses and should be interpreted with caution. Collectively, these data help refine our understanding of which patients may derive the greatest response and further support the role of cabozantinib + nivolumab combination as a first-line therapy option in aRCC.1
Dr Manojkumar Bupathi received a fee for participating in the development of this article, and his comments reflect his opinions and are not intended to constitute medical advice for individual patients.
AXL=AXL receptor tyrosine kinase; BICR=blinded independent central review; CI=confidence interval; HR=hazard ratio; IMDC=International Metastatic RCC Database Consortium; MET=hepatocyte growth factor receptor; NE=not estimable; NR=not reported; ORR=objective response rate; OS=overall survival; PD=progressive disease; PFS=progression-free survival; RECIST=Response Evaluation Criteria in Solid Tumors; VEGFR=vascular endothelial growth factor receptor.
INDICATION
CABOMETYX® (cabozantinib), in combination with nivolumab, is indicated for the first-line treatment of patients with advanced renal cell carcinoma (RCC).
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Hemorrhage: CABOMETYX can cause severe and fatal hemorrhages. The incidence of Grade 3-5 hemorrhagic events was 5% in CABOMETYX patients in RCC, HCC, and DTC studies. Discontinue CABOMETYX for Grade 3-4 hemorrhage and before surgery. Do not administer to patients who have a recent history of hemorrhage, including hemoptysis, hematemesis, or melena.
Perforations and Fistulas: Fistulas, including fatal cases, and gastrointestinal (GI) perforations, including fatal cases, each occurred in 1% of CABOMETYX patients. Monitor for signs and symptoms, and discontinue CABOMETYX in patients with Grade 4 fistulas or GI perforation.
Thromboembolic Events: CABOMETYX can cause arterial or venous thromboembolic events. Venous thromboembolism occurred in 7% (including 4% pulmonary embolism) and arterial thromboembolism in 2% of CABOMETYX patients. Fatal thrombotic events have occurred. Discontinue CABOMETYX in patients who develop an acute myocardial infarction or serious arterial or venous thromboembolic events.
Hypertension and Hypertensive Crisis: CABOMETYX can cause hypertension, including hypertensive crisis. Hypertension was reported in 37% (16% Grade 3 and <1% Grade 4) of CABOMETYX patients. In CABINET (n=195), hypertension occurred in 65% (26% Grade 3) of CABOMETYX patients. Do not initiate CABOMETYX in patients with uncontrolled hypertension. Monitor blood pressure regularly during CABOMETYX treatment. Withhold CABOMETYX for hypertension that is not adequately controlled; when controlled, resume at a reduced dose. Permanently discontinue CABOMETYX for severe hypertension that cannot be controlled with antihypertensive therapy or for hypertensive crisis.
Cardiac Failure: CABOMETYX can cause severe and fatal cardiac failure. Cardiac failure occurred in 0.5% of patients treated with CABOMETYX as a single agent, including fatal cardiac failure in 0.1% of patients. Consider baseline and periodic evaluations of left ventricular ejection fraction. Monitor for signs and symptoms of cardiovascular events. Withhold and resume at a reduced dose upon recovery or permanently discontinue depending on the severity.
Diarrhea: CABOMETYX can cause diarrhea and it occurred in 62% (10% Grade 3) of treated patients. Monitor and manage patients using antidiarrheals as indicated. Withhold CABOMETYX until improvement to ≤ Grade 1; resume at a reduced dose.
Palmar-Plantar Erythrodysesthesia (PPE): CABOMETYX can cause PPE and it occurred in 45% of treated patients (13% Grade 3). Withhold CABOMETYX until PPE resolves or decreases to
Grade 1 and resume at a reduced dose for intolerable Grade 2 PPE or Grade 3 PPE.
Hepatotoxicity: CABOMETYX in combination with nivolumab in RCC can cause hepatic toxicity with higher frequencies of Grades 3 and 4 ALT and AST elevations compared to CABOMETYX alone. With the combination of CABOMETYX and nivolumab, Grades 3 and 4 increased ALT or AST were seen in 11% of patients. Monitor liver enzymes before initiation of treatment and periodically. Consider more frequent monitoring as compared to when the drugs are administered as single agents. Consider withholding CABOMETYX and/or nivolumab, initiating corticosteroid therapy, and/or permanently discontinuing the combination for severe or
life-threatening hepatotoxicity.
Adrenal Insufficiency: CABOMETYX in combination with nivolumab can cause primary or secondary adrenal insufficiency. Adrenal insufficiency occurred in 4.7% (15/320) of patients with RCC who received CABOMETYX with nivolumab, including Grade 3 (2.2%), and Grade 2 (1.9%) adverse reactions. Withhold CABOMETYX and/or nivolumab and resume CABOMETYX at a reduced dose depending on severity.
Proteinuria: Proteinuria was observed in 8% of CABOMETYX patients. Monitor urine protein regularly during CABOMETYX treatment. For Grade 2 or 3 proteinuria, withhold CABOMETYX until improvement to ≤ Grade 1 proteinuria; resume CABOMETYX at a reduced dose. Discontinue CABOMETYX in patients who develop nephrotic syndrome.
Osteonecrosis of the Jaw (ONJ): CABOMETYX can cause ONJ and it occurred in <1% of treated patients. Perform an oral examination prior to CABOMETYX initiation and periodically during treatment. Advise patients regarding good oral hygiene practices. Withhold CABOMETYX for at least 3 weeks prior to scheduled dental surgery or invasive dental procedures. Withhold CABOMETYX for development of ONJ until complete resolution; resume at a reduced dose.
Impaired Wound Healing: CABOMETYX can cause impaired wound healing. Withhold CABOMETYX for at least 3 weeks prior to elective surgery. Do not administer for at least 2 weeks after major surgery and until adequate wound healing. The safety of resumption of CABOMETYX after resolution of wound healing complications has not been established.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS): CABOMETYX can cause RPLS. Perform evaluation for RPLS and diagnose by characteristic finding on MRI any patient presenting with seizures, headache, visual disturbances, confusion, or altered mental function. Discontinue CABOMETYX in patients who develop RPLS.
Thyroid Dysfunction: CABOMETYX can cause thyroid dysfunction, primarily hypothyroidism, and it occurred in 19% of treated patients (0.4% Grade 3). Assess for signs of thyroid dysfunction prior to the initiation of CABOMETYX and monitor for signs and symptoms during treatment.
Hypocalcemia: CABOMETYX can cause hypocalcemia, with the highest incidence in DTC patients. Based on the safety population, hypocalcemia occurred in 13% of CABOMETYX patients (2% Grade 3 and 1% Grade 4).
Monitor blood calcium levels and replace calcium as necessary during treatment. Withhold and resume CABOMETYX at a reduced dose upon recovery or permanently discontinue CABOMETYX depending on severity.
Embryo-Fetal Toxicity: CABOMETYX can cause fetal harm. Advise pregnant women of the potential risk to a fetus and advise females of reproductive potential to use effective contraception during treatment with CABOMETYX and for 4 months after the last dose.
ADVERSE REACTIONS
The most common (≥20%) adverse reactions are:
CABOMETYX in combination with nivolumab: diarrhea, fatigue, hepatotoxicity, PPE, stomatitis, rash, hypertension, hypothyroidism, musculoskeletal pain, decreased appetite, nausea, dysgeusia, abdominal pain, cough, and upper respiratory tract infection.
DRUG INTERACTIONS
Strong CYP3A4 Inhibitors: If coadministration with strong CYP3A4 inhibitors cannot be avoided, reduce the CABOMETYX dosage. Avoid grapefruit or grapefruit juice.
Strong or Moderate CYP3A4 Inducers: If coadministration with strong or moderate CYP3A4 inducers cannot be avoided, increase the CABOMETYX dosage. Avoid St. John’s wort.
USE IN SPECIFIC POPULATIONS
Lactation: Advise women not to breastfeed during CABOMETYX treatment and for 4 months after the final dose.
Hepatic Impairment: In patients with moderate hepatic impairment, reduce the CABOMETYX dosage. Avoid CABOMETYX in patients with severe hepatic impairment.
Please see accompanying full Prescribing Information by clicking here.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.
References
- Suárez C, Motzer RJ, Powles T, et al. Characterization of depth and durability of response in patients with previously untreated advanced renal cell carcinoma who received cabozantinib plus nivolumab: long-term follow-up and exploratory analysis of CheckMate 9ER. Poster Presented at: American Society of Clinical Oncology Genitourinary Cancers Symposium. February 26-28, 2026.
- Zarrabi KK, Lanade O, Geynisman DM. Determining front-line therapeutic strategy for metastatic clear cell renal cell carcinoma. Cancers. 2022;14;4607. doi.org/10.3390/cancers14194607.
- Choueiri TK, Powles T, Burotto M, et al; CheckMate 9ER Investigators. Nivolumab plus cabozantinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med. 2021;384(9):829‐841.
- CABOMETYX® (cabozantinib) Prescribing Information. Exelixis, Inc.
- Motzer RJ, Escudier B, Burotto M, et al. Final analysis of nivolumab plus cabozantinib for advanced renal cell carcinoma from the randomized phase III CheckMate 9ER trial. Ann Oncol. 2025; Published online September 23, 2025. doi:10.1016/j.annonc.2025.09.006.
- Powles T, Choueiri TK, Burotto M, et al. Final overall survival analysis and organ-specific target lesion assessments with 2-year follow-up in CheckMate 9ER: nivolumab plus cabozantinib versus sunitinib for patients with advanced renal cell carcinoma. Poster presented at: American Society of Clinical Oncology Genitourinary Cancers Symposium; February 17-19, 2022.
- Data on file. Exelixis, Inc.
©2026 Exelixis, Inc. CA-3913 04/26
OPDIVO® and the related logo are registered trademarks of Bristol‐Myers Squibb Company.
