SUMMARY: The FDA on May 26, 2020, approved the combination of OPDIVO® (Nivolumab) plus YERVOY® (Ipilimumab) and 2 cycles of Platinum-doublet chemotherapy as first-line treatment for patients with metastatic or recurrent Non-Small Cell Lung Cancer (NSCLC), with no Epidermal Growth Factor Receptor (EGFR) or Anaplastic Lymphoma Kinase (ALK) genomic tumor aberrations. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the immune system T cells. Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152) and PD-1(Programmed cell Death 1). Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response. OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4.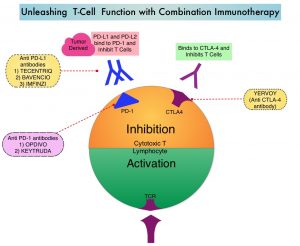
In the CheckMate-227, Part 1, Phase III trial, a combination of OPDIVO® plus YERVOY® significantly improved Overall Survival (OS), Progression Free Survival (PFS), Objective Response Rates (ORR) and Duration of Response, compared to chemotherapy, independent of PD-L1 expression level. The authors in this study hypothesized that a limited course of chemotherapy combined with OPDIVO® plus YERVOY® could provide rapid disease control, while building on the durable Overall Survival benefit seen with dual PD-1 and CTLA-4 inhibition.
CheckMate-9LA is a randomized, open-label, multi-center, Phase III trial which evaluated the benefit of a combination of OPDIVO® plus YERVOY®, and 2 cycles of Platinum-doublet chemotherapy versus Platinum-doublet chemotherapy for 4 cycles followed by optional Pemetrexed maintenance therapy, as a first-line treatment in patients with metastatic or recurrent NSCLC, regardless of PD-L1 status and histology. In this study, 719 adults treatment naïve patients with histologically confirmed Stage IV/recurrent NSCLC, with ECOG Performance Status 0-1, and no known sensitizing EGFR/ALK alterations, were randomly assigned 1:1 to receive OPDIVO® 360 mg every 3 weeks plus YERVOY® 1 mg/kg every 6 weeks and 2 cycles of platinum-doublet chemotherapy (N=361), or 4 cycles of platinum-doublet chemotherapy alone (N=358). Chemotherapy was based on histology. Patients with non-squamous NSCLC in the chemo-only randomized group could receive optional Pemetrexed maintenance treatment. Patients were treated with immunotherapy until disease progression, unacceptable toxicity, or for 2 years. Patients were stratified by PD-L1 status (less than 1% versus 1% or more), sex, and histology (squamous versus non-squamous). Demographics in treatment groups were well balanced. The Primary end point was Overall Survival (OS). Secondary endpoints included Progression Free Survival (PFS), Objective Response Rate (ORR) and efficacy by PD-L1 subgroups.
At a preplanned interim analysis after a minimum follow up 8.1 months, this trial demonstrated a statistically significant benefit in OS for patients treated with OPDIVO® plus YERVOY® and limited chemotherapy, compared to those who received chemotherapy alone. The median OS was 14.1 months versus 10.7 months, respectively (HR=0.69; P=0.0006). With longer follow up at 12.7 months, this OS benefit continued to improve in the immunotherapy plus chemotherapy group, with a median OS of 15.6 months versus 10.9 months, respectively (HR=0.66). The 1-year OS rates were 63% versus 47%. This clinical benefit was consistent across all efficacy measures in key subgroups including by PD-L1 and histology.
The median PFS was 6.8 months in the OPDIVO® plus YERVOY® and chemotherapy group and 5 months in the chemotherapy-only group (HR=0.70; P=0.0001). The ORR was 38% and 25%, respectively (P= .0003). The median response duration was 10 months in the OPDIVO® plus YERVOY® and chemotherapy group, and 5.1 months in the chemotherapy-only group. Grade 3-4 treatment related Adverse Events were reported in 47% of the patients receiving the immunotherapy plus chemotherapy combination versus 38% in the chemotherapy-only group.
It was concluded that CheckMate 9LA met its Primary endpoint of Overall Survival, and OPDIVO® plus YERVOY® with a limited course of chemotherapy should be considered as a new first line treatment option for patients advanced Non Small Cell Lung Cancer.
Nivolumab (NIVO) + ipilimumab (IPI) + 2 cycles of platinum-doublet chemotherapy (chemo) vs 4 cycles chemo as first-line (1L) treatment (tx) for stage IV/recurrent non-small cell lung cancer (NSCLC): CheckMate 9LA. Reck M, Ciuleanu T-E, Dols MC, et al. J Clin Oncol 38: 2020 (suppl; abstr 9501)