
SUMMARY: It is estimated that in the United States approximately 13,000 people are diagnosed with MyeloDysplastic Syndromes (MDS) each year. MyeloDysplastic Syndromes are a heterogenous group of stem cell disorders characterized by marrow failure resulting in cytopenias with associated cytogenetic abnormalities, and abnormal cellular maturation with morphologic changes in clonal cells. Majority of the individuals diagnosed with MDS are aged 65 years and older and die as a result of infection and/or bleeding consequent to bone marrow failure. About a third of patients with MDS develop Acute Myeloid Leukemia (AML).
Patients with Lower-risk MDS (Revised IPSS-Very Low, Low, or Intermediate risk ) often present with symptomatic anemia and these patients are in chronic need for RBC transfusions which in turn can result in iron overload and can have a negative impact on quality of life and Overall Survival. These patients are treated with Erythropoiesis Stimulating Agents (ESAs) as first line therapy. ESAs such as Darbepoetin alfa and Epoetin alfa are re-engineered and recombinant DNA technology products of Erythropoietin (EPO), and they stimulate erythropoiesis by binding and activating the EPO receptor. However, transfusion-dependent patients with serum EPO levels above 200 U per liter are less likely to respond to ESAs. Additionally, patients with MDS with ring sideroblasts have a shorter median duration of response to ESAs, than those who do not have ring sideroblasts. Patients with Lower-risk MDS with chromosome 5q deletion (del 5q) who are transfusion dependent are treated with Lenalidomide, regardless of previous treatment with ESAs. In contrast, only 39% of patients with non-del(5q) Lower-risk MDS receive second line therapy besides RBC transfusions, and there are few treatment options for patients who are refractory to, unresponsive to, or ineligible for ESAs. There is therefore an unmet clinical need for safe and effective treatment options, to reduce the RBC transfusion burden in these patients.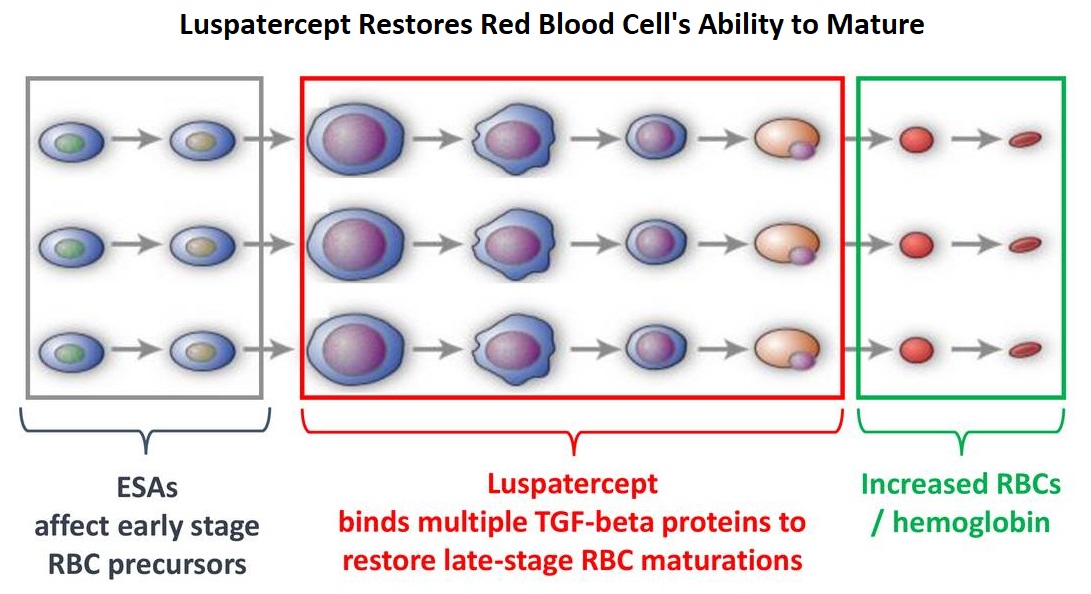
Signaling by the SMAD2 and SMAD3 pathway exerts an inhibitory effect on red cell maturation. This pathway is constitutively activated in the bone marrow cells of patients with MDS and diseases associated with ineffective erythropoiesis such as β-thalassemia. REBLOZYL® (Luspatercept) is a recombinant soluble fusion protein and is first-in-class erythroid maturation agent that enhances erythropoiesis by promoting late-stage Red Blood Cell precursor differentiation and maturation. It targets select Transforming Growth Factor (TGF)-β superfamily ligands such as GDF11, that regulate late-stage erythropoiesis. This results in a reduction in aberrant SMAD2 and SMAD3 signaling, thereby promoting late-stage RBC precursor differentiation and maturation. In a Phase II study, treatment of Lower-risk MDS patients with REBLOZYL® resulted in 38% of patients being transfusion independent for 8 weeks or longer and this benefit was even more so among patients with 15% or more ring sideroblasts.
The MEDALIST trial is a randomized, double-blind, placebo-controlled Phase III study which evaluated the efficacy and safety of REBLOZYL® in patients with anemia secondary to MDS, defined as Very Low-Risk, Low-Risk, or Intermediate-Risk with Ring Sideroblasts, according to the Revised International Prognostic Scoring System (R-IPSS). Eligible patients were refractory, intolerant, or ineligible to receive ESAs and required RBC transfusions. A total of 229 patients (N=229) were randomized 2:1 to receive either REBLOZYL® at a starting dose level of 1mg/kg SC with titration up to 1.75 mg/kg if needed (N=153), or placebo SC (N=76), every 3 weeks for 24 weeks or more. The median age was 71 years and median time from diagnosis was 41.8 months. Approximately 95% of patients had previously received ESAs and 90% had an SF3B1 mutation. The Primary endpoint was RBC transfusion independence for 8 weeks or more between week 1 and 24. A key Secondary endpoint was RBC transfusion independence for 12 weeks or more between week 1 and 24.
Among those receiving REBLOZYL®, 38% achieved the Primary endpoint of RBC transfusion independence for 8 weeks or more, compared with 13% receiving placebo (P<0.001). Further among those receiving REBLOZYL®, 28% achieved the key Secondary endpoint of RBC transfusion independence for 12 weeks or more compared with 8% receiving placebo (P<0.001). The median duration of the longest, single continuous period of response to REBLOZYL® was 30.6 weeks, and 13.6 weeks in the placebo group. Among patients who had a baseline transfusion burden of 4 to less than 6 units per 8 weeks, 37% of those in the REBLOZYL® group and 4% of those in the placebo group had a response. Additionally, patients receiving REBLOZYL® were more likely to achieve an mHI-E (modified Hematologic Improvement-Erythroid) response, (defined as a reduction in transfusion of 4 or more RBC units per 8 weeks or a mean hemoglobin increase of 1.5 g/dL or more per 8 weeks, in the absence of transfusions), compared with patients receiving placebo (53% versus 12% during weeks 1-24; P<0.0001). A mean increase in hemoglobin level of at least 1 g/dL during weeks 1 to 24 was noted in 35% of patients who received REBLOZYL® and in 8% of patients who received placebo. The most common adverse events of any grade associated with REBLOZYL® included fatigue, diarrhea, asthenia, nausea and dizziness, and the incidence of adverse events decreased over time.
It was concluded that treatment with REBLOZYL® significantly reduced the severity of anemia in patients with Lower-risk MDS with ring sideroblasts, who had been RBC transfusion-dependent, and who had disease that was refractory to or unlikely to respond to ESAs. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. Fenaux P, Platzbecker U, Mufti GJ, et al. N Engl J Med 2020; 382:140-151
Author: RR
Neoadjuvant KEYTRUDA® Plus Chemotherapy Improves Pathological Complete Response in Triple Negative Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (13%) will develop invasive breast cancer during their lifetime. Approximately 276,480 new cases of invasive female breast cancer will be diagnosed in 2020 and about 42,170 women will die of the disease.
Triple Negative Breast Cancer (TNBC) is a heterogeneous, molecularly diverse group of breast cancers and are ER (Estrogen Receptor), PR (Progesterone Receptor) and HER2 (Human Epidermal Growth Factor Receptor-2) negative. TNBC accounts for 15-20% of invasive breast cancers, with a higher incidence noted in young patients. It is usually aggressive, and tumors tend to be high grade and patients with TNBC are at a higher risk of both local and distant recurrence. Those with metastatic disease have one of the worst prognoses of all cancers with a median Overall Survival of 13 months. The majority of patients with TNBC who develop metastatic disease do so within the first 3 years after diagnosis, whereas those without recurrence during this period of time have survival rates similar to those with ER-positive breast cancers. The lack of known recurrent oncogenic drivers in patients with metastatic TNBC, presents a major therapeutic challenge. Nonetheless, patients with TNBC often receive chemotherapy in the neoadjuvant, adjuvant or metastatic settings and approximately 30-40% of patients achieve a pathological Complete Response (pCR) in the neoadjuvant setting. In addition to increasing the likelihood of tumor resectability and breast preservation, patients achieving a pCR following neoadjuvant chemotherapy have a longer Event Free Survival (EFS) and Overall Survival (OS). Those who do not achieve a pathological Complete Response tend to have a poor prognosis. For all these reasons, pCR is considered a valid endpoint for clinical testing of neoadjuvant therapy in patients with early stage TNBC. It appears that there are subsets of patients with TNBC who may be inherently insensitive to cytotoxic chemotherapy. Three treatment approaches appear to be promising and they include immune therapies, PARP inhibition and inhibition of PI3K pathway. Previously published studies have shown that presence of tumor-infiltrating lymphocytes was associated with clinical benefit, when treated with chemotherapy and immunotherapy, in patients with TNBC, and improved clinical benefit was observed in patients with immune-enriched molecular subtypes of metastatic TNBC.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. Cytotoxic chemotherapy releases tumor-specific antigens and immune checkpoint inhibitors such as KEYTRUDA® when given along with chemotherapy can enhance endogenous anticancer immunity. Preliminary results from Phase I and II trials have shown that in patients with TNBC, KEYTRUDA® given along with chemotherapy in a neoadjuvant setting resulted in a high rate of pCR.
KEYNOTE-522 is an international, placebo controlled Phase III trial, conducted to evaluate the safety and efficacy of neoadjuvant KEYTRUDA® plus chemotherapy followed by adjuvant KEYTRUDA® or placebo, in patients with early stage TNBC. In this study, 1,174 patients were randomly assigned in a 2:1 ratio to receive neoadjuvant KEYTRUDA® 200 mg IV every 3 weeks (N=784) or placebo (N=390). All patients received 4 cycles of Carboplatin plus Paclitaxel, followed by 4 cycles of Doxorubicin or Epirubicin plus Cyclophosphamide, in the neoadjuvant setting. Following definitive surgery, adjuvant KEYTRUDA® or placebo was continued every 3 weeks for 9 cycles or until disease recurrence or unacceptable toxicity. Enrolled TNBC patients were newly diagnosed, treatment naïve, and included both node-negative and node-positive patients with nonmetastatic disease (Tumor Stage T1c, Nodal Stage N1-N2 or Tumor Stage T2-T4, Nodal Stage N0-N2, per AJCC criteria). Treatment groups were well balanced and patients were stratified according to nodal status, tumor size, and Carboplatin schedule (weekly versus every 3 weeks). The two Primary endpoints were pathological Complete Response (pCR) at the time of definitive surgery and Event Free Survival (EFS). The median follow up was 15.5 months.
At the first interim analysis, the pCR among the first 602 patients who underwent randomization was 64.8% in the KEYTRUDA® plus chemotherapy group, compared with 51.2% in the placebo plus chemotherapy group (P<0.001). This pCR benefit was consistent across subgroups including PD-L1 expresssion subgroups. In the PD-L1-positive population, the pCR was 68.9% in the KEYTRUDA® plus chemotherapy group compared with 54.9% in the placebo plus chemotherapy group. In the PD-L1 negative group, the pCR in the KEYTRUDA® plus chemotherapy group was 45.3% and 30.3% in the placebo plus chemotherapy group. Neoadjuvant KEYTRUDA® plus chemotherapy followed by adjuvant KEYTRUDA® showed a favorable trend for Event Free Survival compared with chemotherapy alone, although these data are still premature. Across all treatment phases, Grade 3 or higher treatment-related toxicities were 78.0% in the KEYTRUDA® plus chemotherapy group and 73.0% in the placebo plus chemotherapy group.
It was concluded that among patients with early stage Triple Negative Breast Cancer, the addition of KEYTRUDA® to neoadjuvant chemotherapy significantly increased the pathological Complete Response rate, compared to those who received placebo plus neoadjuvant chemotherapy, with a favorable trend in Event Free Survival. Pembrolizumab for Early Triple-Negative Breast Cancer. Schmid P, Cortés J, Pusztai L, et al. for the KEYNOTE-522 Investigators. N Engl J Med 2020;382:810-821
Late Breaking Abstract – ASH 2019: DARZALEX®, KYPROLIS® and Dexamethasone Combination Improves PFS in Relapsed or Refractory Multiple Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 32, 270 new cases will be diagnosed in 2020 and 12,830 patients are expected to die of the disease. Multiple Myeloma (MM) in 2020 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Multiple Myeloma is a disease of the elderly, with a median age at diagnosis of 69 years and characterized by intrinsic clonal heterogeneity. Almost all patients eventually will relapse, and patients with a high-risk cytogenetic profile or refractory disease have the worst outcomes. The median survival for patients with Myeloma is over 10 years.
REVLIMID® (Lenalidomide) in combination with VELCADE® (Bortezomib) and Dexamethasone is the preferred regimen according to the NCCN guidelines, for both transplant and non-transplant candidates with newly diagnosed Multiple Myeloma, and when given continuously or with maintenance therapy, has improved survival outcomes. Nonetheless, a significant number of patients progress while on these agents or discontinue therapy due to toxicities. There is therefore a need for effective and tolerable regimens for patients who are exposed or refractory to REVLIMID® or VELCADE®.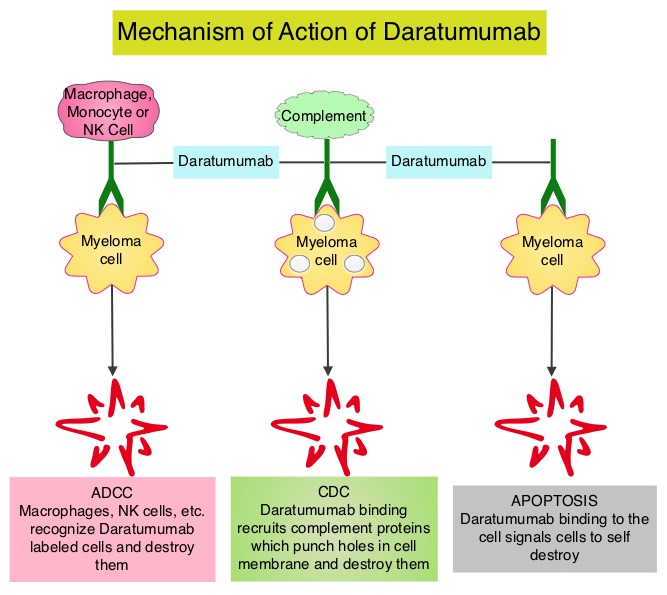
KYPROLIS® (Carfilzomib) is a second generation selective, epoxyketone Proteasome Inhibitor and unlike VELCADE®, proteasome inhibition with KYPROLIS® is irreversible. DARZALEX® (Daratumumab) is a human IgG1 antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Dependent Cytotoxicity (CDC) and direct Apoptosis. Additionally, DARZALEX® may play a role in immunomodulation, by depleting CD38-positive regulator immune suppressor cells, and thereby expanding T cells, in patients responding to therapy. Both KYPROLIS® and DARZALEX® are approved as single agents, as well as in combination with other drugs, for the treatment of patients with Relapsed/Refractory Multiple Myeloma. In a Phase I study, KYPROLIS® in combination with Dexamethasone and DARZALEX® demonstrated safety and efficacy in patients Relapsed/Refractory Multiple Myeloma.
CANDOR is a multicenter, open-label, Phase III trial, which included Relapsed/Refractory Multiple Myeloma patients with measurable disease who had received 1-3 prior lines of therapy, with Partial Response or better to one or more lines of therapy. A total of 466 patients were randomly assigned 2:1 to receive triplet of KYPROLIS®, Dexamethasone, and DARZALEX® (KdD)- N=312 or KYPROLIS® and Dexamethasone (Kd) alone- N=154. All patients received KYPROLIS® as a 30 minute IV infusion on days 1, 2, 8, 9, 15, and 16 of each 28-day cycle (20 mg/m2 on days 1 and 2 during cycle 1 and 56 mg/m2 thereafter). DARZALEX® 8 mg/kg was administered IV on days 1 and 2 of cycle 1 and at 16 mg/kg once weekly for the remaining doses of the first 2 cycles, then every 2 weeks for 4 cycles (cycles 3-6), and every 4 weeks thereafter. All patients received Dexamethasone 40 mg oral or IV weekly (20 mg for patients over 75 years of age). The median age was 64 years, 42% and 90% received prior REVLIMID® and VELCADE® (Bortezomib) containing regimens respectively, and a third of patients were refractory to REVLIMID®. The Primary endpoint was Progression Free Survival (PFS) and Secondary endpoints including Overall Response Rate (ORR), Minimal Residual Disease (MRD)-negative status, Complete Response (CR) rate at 12 months, Overall Survival (OS), Duration of Response, and Safety.
After a median follow up of 17 months, the study met its Primary endpoint and the median PFS was not reached for the KdD arm and was 15.8 months for the Kd arm (HR=0.63; P=0014). This represented a 37% reduction in the risk of progression or death in the KdD group. The PFS benefit of KdD was maintained across prespecified subgroups, particularly among REVLIMID®-exposed and REVLIMID®-refractory patients. The ORR was 84.3% in the KdD group versus 74.7% in the Kd group (P=0.004), with a CR rate or better of 28.5% versus 10.4% respectively. The median time to first response was one month in both treatment groups. Patients treated with KdD achieved deeper responses which was nearly 10 times higher, with a MRD-negative Complete Response rate at 12 months of 12.5% for KdD versus 1.3% for Kd (P<0.0001). The median treatment duration was longer in the KdD group compared to the Kd group (70.1 versus 40.3 wks). The median OS was not reached in either groups, at a median follow up time of 17 months. Toxicities were generally manageable and the incidence of Adverse Events leading to treatment discontinuation was similar in both treatment groups.
It was concluded that a combination of KYPROLIS® along with Dexamethasone and DARZALEX® resulted in a significant PFS benefit over KYPROLIS® and Dexamethasone alone, with deeper responses, and the PFS benefit of KdD was maintained across prespecified, clinically important subgroups, particularly REVLIMID®-exposed and REVLIMID®-refractory patients. The authors added that KdD regimen should be considered as a novel, efficacious, and tolerable immunomodulatory-free treatment option for Relapsed/Refractory Multiple Myeloma patients. Carfilzomib, Dexamethasone, and Daratumumab Versus Carfilzomib and Dexamethasone for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma (RRMM): Primary Analysis Results from the Randomized, Open-Label, Phase 3 Study Candor (NCT03158688). Usmani SZ, Quach H, Mateos M-V, et al. Presented at the 61st American Society of Hematology Annual Meeting and Exposition; Orlando, Florida; December 7-10, 2019; Abstract LBA-6.
DERMATOLOGIC TOXICITIES OF CANCER TREATMENT
Coming Soon!
CHEMOTHERAPY INDUCED ANEMIA
Coming Soon!
MYELOID GROWTH FACTORS
DEFINITIONS: Neutropenia is defined as an Absolute Neutrophil Count (ANC) of 500 or less per microL or ANC less than 1000/microL and a predicted decline to 500 or less per microL over the next 48 hours. Febrile Neutropenia (FN) is defined as a temperature of 38.0 degrees C orally in a Neutropenic patient.
RISK FACTORS: The risk for of Febrile Neutropenia is not only dependent on the treatment regimen and delivered dose intensity but also on the overall condition of the patient. It is important that all these risk factors be taken into consideration before considering Primary Prophylaxis with CSF (Colony Stimulating Factors given prior to first chemotherapy cycle).
Patients at a risk of developing Febrile Neutropenia include
1) Patients 65 years of age or older
2) Poor Performance Status
3) Previous chemotherapy or radiation therapy
4) Preexisting Neutropenia
5) Tumor involving the bone marrow
6) Poor Liver function with elevated bilirubin
7) Poor renal function
8) Recent surgery
9) Preexisting Infection
10) Open wounds
Based on the treatment regimen and patient related risk factors, patients can fall in one of the three risk groups to develop Febrile Neutropenia (FN)
High Risk: More than 20% risk of FN
Intermediate Risk: 10-20% risk of FN
Low Risk: Less than 10% risk
Approximately 25%-40% of patients who had no prior chemotherapy develop FN with commonly prescribed chemotherapy regimens1. A notable group falling in the high risk category are those in whom dose intensity has to be maintained, in spite of a Neutropenic complication in the immediate previous chemotherapy cycle. The risk of FN associated with various chemotherapy regimens has been extensively published2 and is beyond the scope of this review.
BENEFITS OF CSF:
1) Use of G-CSF (Granulocyte – Colony Stimulating Factor) as recommended, reduces the incidence, severity and duration of chemotherapy induced Neutropenia. Neutropenic colitis or Typhlitis can be associated with prolonged neutropenia in patients with hematologic malignancies. The decreased duration of WBC (White Blood Cell) nadir in turn can decrease the chance of infectious complications including infection related mortality and improve quality of life3
2) There can be potential cost savings, preventing patient hospitalization4
3) G-CSF can facilitate delivery of full dose intensity of chemotherapy on schedule5
RISKS ASSOCIATED WITH CSF:
1) Mild to moderate bone pain
2) Rare cases of Splenic rupture
3) Allergic reactions involving the skin, cardiovascular and respiratory system with Filgrastim
FDA APPROVED MYELOID GROWTH FACTORS:
NEUPOGEN® (Filgrastim)
NEULASTA® (Pegfilgrastim)
NEUTROVAL® (Tbo-filgrastim)
LEUKINE® (Granulocyte Macrophage-CSF, Sargramostim)
The first three are currently approved for use in preventing chemotherapy induced Neutropenia, whereas LEUKINE® is indicated for use in patients with Acute Myeloid Leukemia following induction therapy and in Stem Cell Transplantation
PREVENTION OF NEUTROPENIC COMPLICATIONS:
1) Primary prophylaxis (Prior to start of the first cycle of chemotherapy) with CSF’s is recommended for all patients who are considered to be at high risk for FN6, regardless of whether the treatment intent is palliative or curative
2) CSF’s should be considered on an individualized basis for patients who fall in the Intermediate risk category.
3) CSF’s are not usually recommended for patients considered to be at low risk for FN. The exception however will be for those patients who are being treated with a curative intent or receiving adjuvant therapy who cannot risk FN as this could lead to fatal complications.
4) Research in the area of pharmacogenomics may help us better understand how Neutropenic complications could be prevented
NEUTROPENIA AFTER FIRST CYCLE OF CHEMOTHERAPY:
1) FN or dose limiting Neutropenia (Unable to give treatment on schedule) following first cycle of chemotherapy warrants use of CSF
2) If a patient experiences dose limiting Neutropenia or FN inspite of primary prophylaxis with CSF’s, dose reductions or change in treatment regimen has to be considered
3) For a patient hospitalized with FN, CSF’s may be considered if the patient is deemed to be at high risk, provided the patient did not receive prophylactic NEULASTA® following chemotherapy. NEULASTA® is only indicated for the prevention of chemotherapy induced Neutropenia but not for therapy, when a patient presents with Neutropenia.
ADMINISTRATION OF CSF:
1) Start NEUPOGEN® within 24-72 hours after completion of chemotherapy and continue until post nadir ANC is normal or close to normal
2) NEULASTA® is administered 24 hours after completion of chemotherapy and is not recommended for administration on a schedule less than 2 weeks.
3) NEULASTA® is not recommended following weekly chemotherapy2.
MANAGEMENT of Febrile Neutropenia – OTHER ASPECTS:
1) Hospitalization is recommended
2) Patients should be started on broad spectrum antibiotics, following appropriate cultures
3) Start CSF if appropriate
4) Good hand hygiene
5) If prolonged Neutropenia is anticipated, minimize exposure to pets and live plants
Reference List
1. Dale DC. Colony-stimulating factors for the management of neutropenia in cancer patients. Drugs 2002;62 Suppl 1:1-15.
2. Crawford J, Allen J, Armitage J et al. Myeloid growth factors. J Natl Compr Canc Netw 2011;9(8):914-932.
3. Fortner BV, Schwartzberg L, Tauer K, Houts AC, Hackett J, Stolshek BS. Impact of chemotherapy-induced neutropenia on quality of life: a prospective pilot investigation. Support Care Cancer 2005;13(7):522-528.
4. Lyman GH, Kuderer NM. The economics of the colony-stimulating factors in the prevention and treatment of febrile neutropenia. Crit Rev Oncol Hematol 2004;50(2):129-146.
5. Citron ML, Berry DA, Cirrincione C et al. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol 2003;21(8):1431-1439.
6. Smith TJ, Khatcheressian J, Lyman GH et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 2006;24(19):3187-3205.
PAIN MANAGEMENT
Coming Soon!
CHEMOTHERAPY INDUCED NAUSEA AND VOMITING (CINV)
Chemotherapy Induced Nausea and Vomiting (CINV) is quite common and occurs in about 80% of patients receiving chemotherapy1,2. It is important to differentiate nausea from vomiting, as more patients experience nausea rather than vomiting3. There are 6 different categories of CINV
1. Acute CINV
2. Delayed CINV
3. Breakthrough CINV
4. Refractory CINV
5. Anticipatory CINV
6. Radiation Induced Nausea and Vomiting
ACUTE CINV: Acute CINV by definition begins within the first 24 hours following chemotherapy administration, with most patients experiencing symptoms within the first four hours of treatment.
DELAYED CINV: This is defined as nausea and vomiting occurring more than 24 hours after chemotherapy administration and can persist for several days4. This is often underestimated, as a third of the patients receiving chemotherapy may experience delayed nausea and vomiting with out prior acute nausea or vomiting.
BREAKTHROUGH CINV: This is defined as nausea and/or vomiting experienced by patients despite prophylactic treatment with antiemetics. These patients require additional intervention with antiemetics to treat their symptoms.
REFRACTORY CINV: Patients on chemotherapy may experience nausea and vomiting following subsequent cycles of chemotherapy administration when prophylaxis/rescue for nausea and vomiting have failed in earlier cycles.
ANTICIPATORY CINV: This is a “learned conditioned response” with psychologic undertones as a result of prior experience of nausea and vomiting with chemotherapy. It usually occurs about 12 hours leading up to chemotherapy administration5,6. The incidence varies from 20%-50%. It is more often seen in younger patients and nausea is more common than vomiting. This entity is difficult to treat and may require psychologic counseling.
RADIATION INDUCED NAUSEA AND VOMITING: This is often experienced by patients undergoing Total Body Irradiation (TBI), a technique that is used prior to bone marrow transplantation7. Patients undergoing radiation to the upper abdomen will also experience nausea and vomiting8.
Pathophysiology of Nausea and Vomiting
The brain controls the multistep pathway that results in vomiting. There are two separate units in the medulla that play a vital role in the causation of nausea and vomiting.
• Chemoreceptor trigger zone (CTZ) also called Area Postrema, present on the floor of the fourth ventricle. This area is very sensitive to chemical stimuli and is easily accessible to emetogenic substances in the general circulation.
• Vomiting Center (Emetic Center) located in the reticular formation of the medulla. This center integrates the emetic response and coordinates the act of vomiting.
The neuroreceptors involved in mediating vomiting include Serotonin (5HT), Dopamine (D2), Neurokinin-1(NK-1), Cannabinoid(CB1), Opiate, Histamine(H1), Corticosteroid and Acetylcholine or Muscarinic(M) receptors. These receptors are located in the vomiting and vestibular centers of the brain9. The most important receptors involved in the emetic response however are Serotonin and Dopamine receptors10. Vomiting is triggered by the Vomiting Center after it receives impulses from CTZ, GI tract, and cerebral cortex and vestibular apparatus in the inner ear. Chemotherapeutic agents and radiation therapy generally induce vomiting by producing free radicals, which in turn act on the enterochromaffin cells, resulting in the release of Serotonin(5-hydroxytryptamine-5HT3). Serotonin activates the serotonin receptors. Activation of the receptors then activates the vagal afferent pathway, which in turn activates the vomiting center and causes an emetic response. Chemotherapeutic agents along with its metabolites also directly stimulate the CTZ by way of general circulation, triggering vomiting through the Vomiting Center.
Classification of Chemotherapeutic agents based on Emetogenicity11
High Risk: More than 90% of the patients experience acute emesis
Moderate Risk: 30-90% of the patients experience acute emesis
Low Risk: 10-30% of the patients experience acute emesis
Minimal Risk: Less than 10% of the patients experience acute emesis
Risk Factors for Nausea and Vomiting
• Emetogenic potential of the chemotherapy agent used
• Younger age
• Female gender
• History of motion sickness
• Alcohol consumption
Classification of Antiemetics
Serotonin (5-HT3) Receptor Antagonists
ZOFRAN ® (Ondansetron): This agent is the first in its class approved by the FDA in 1991 for the treatment of CINV. This first generation 5-HT3 receptor antagonist is available as an IV preparation, sublingual/chewable tablet, oral tablet and oral solution. This agent has a shorter half life than KYTRIL® and ALOXI®.
KYTRIL® (Granisetron): This first generation 5-HT3 receptor antagonist provides 24-hour protection against chemotherapy induced nausea and vomiting. It is available as a single dose Injection as well as tablets or oral solution
ALOXI ® (Palonosetron): This second generation 5-HT3 antagonist has a 100 fold higher binding affinity to 5-HT3 receptor than other 5-HT3 receptor antagonists12. This agent is effective in preventing both acute and delayed onset nausea and vomiting. This agent has to be given IV and oral administration is not feasible due to poor bioavailability, unlike the first generation 5-HT3 antagonists.
Neurokinin-1 (NK-1) Receptor Antagonist
EMEND® (Aprepitant): This agent selectively blocks the binding of substance P to the NK-1 receptor in the central nervous system and thereby complements the antiemetic activity of 5-HT3 receptor antagonists by virtue of its different mechanism of action. It is available as tablets as well as parenteral preparation. This agent can interact with several other drugs more, so when given orally because of first-pass metabolism13. It is therefore important to check the package insert for drug interactions.
Dopamine Receptor Antagonists
These agents antagonize dopamine (D2)-receptors which are involved in the emetic signaling through the chemoreceptor trigger zone. Dopamine receptor antagonists also counteract dopamine receptors in the stomach, implicated in decreasing stomach motility during nausea and vomiting. The three main classes of dopamine receptor antagonists are phenothiazines, butyrophenones, and benzamides.
Phenothiazines – PHENERGAN® (promethazine) belongs to this class. Low doses of phenothiazines antagonize interaction of dopamine with D2-receptors and thus exert an antiemetic effect.
Benzamides – Are strong central and peripheral D2-antagonists. They exert antiemetic effects by increasing lower esophageal sphincter tone and decreasing transit time through the upper gastrointestinal tract. REGLAN® (metoclopramide) belongs to this class.
Butyrophenones
HALDOL® (Haloperidol) belongs to this group of agents.
H1 Receptor Antagonists
Antihistamines antagonize the H1 receptors and inhibit the action of histamine. They also affect the vestibular system, decreasing stimulation of the vomiting center. Further, they also exhibit activity by inhibiting the muscarinic receptor. However, second generation antihistamines such as CLARITIN® (Loratidine), ALLEGRA® (Fexofenadine) and ZYRTEC® (Cetirizine) do not cross the blood brain barrier, and as such do not cause drowsiness and cannot be used as antiemetics. Some examples of H1 receptor antagonists include
• DRAMAMINE® (Dimenhydrinate)
• ANTIVERT® (Meclizine)
• BENADRYL® (Diphenhydramine)
Muscarinic Receptor Antagonists
These agents are good for motion sickness. They antagonize the acetylcholine receptors in the brain. Scopolamine transdermal belongs to this class
Cannabinoids Receptor Antagonists
These agents antagonize the CB1 receptors in the brain. The two drugs in this class
• MARINOL® (Dronabinol)
• CESAMET® (Nabilone)
Corticosteroids
The mechanism of action not clear. It may be related to the inhibition of arachidonic acid release. The agents in this class include
• DECADRON® (Dexamethasone)
• SOLUMEDROL® (Methylprednisolone)
Benzodiazepines
These agents are sometimes effective for anticipatory nausea and vomiting associated with cancer therapy. It may also be useful for vestibular disorders.
• VALIUM® (Diazepam)
• ATIVAN® (Lorazepam)
• XANAX®: (Alprazolam) Miscellaneous
• TIGAN® (Trimethobenzamide)
General Principles of treating Nausea and Vomiting
1) Prevention is better than cure. Aggressively preventing nausea and vomiting will not only improve patients quality of life but will also decrease the incidence of anticipatory nausea and vomiting.
2) The addition of DECADRON® to 5-HT3 and NK-1 receptor antagonists improves the efficacy of the antiemetic regimen.
3) The choice of an antiemetic regimen should be based on patient risk factors as well as emetogenic potential of a given chemotherapy regimen.
4) The toxicity of a given antiemetic agent and potential drug interactions should be taken into consideration before prescribing.
5) Breakthrough emesis should be aggressively managed with round the clock dosing rather than PRN dosing and a drug from a different class should be considered for treatment of this entity. Also consider rectal or IV route of administration rather than PO route.
6) Ensure patients are well hydrated and electrolyte imbalances are promptly addressed.
7) Think “outside the box” when addressing nausea and vomiting in cancer patients. Do not overlook bowel obstruction, brain metastases, uremia, nausea from pain meds such as opiates, chemo or tumor related gastroparesis and anticipatory nausea and vomiting.
8) Anticipatory nausea and vomiting can be difficult to treat and benzodiazepines (ATIVAN®, XANAX®) along with behavioral therapy may be beneficial.
Reference List
1. Morran C, Smith DC, Anderson DA, McArdle CS. Incidence of nausea and vomiting with cytotoxic chemotherapy: a prospective randomised trial of antiemetics. Br Med J 1979; 1(6174):1323-1324.
2. Jenns K. Importance of nausea. Cancer Nurs 1994; 17(6):488-493.
3. Hickok JT, Roscoe JA, Morrow GR et al. 5-Hydroxytryptamine-receptor antagonists versus prochlorperazine for control of delayed nausea caused by doxorubicin: a URCC CCOP randomised controlled trial. Lancet Oncol 2005; 6(10):765-772.
4. Kris MG, Gralla RJ, Clark RA et al. Incidence, course, and severity of delayed nausea and vomiting following the administration of high-dose cisplatin. J Clin Oncol 1985; 3(10):1379-1384.
5. Moher D, Arthur AZ, Pater JL. Anticipatory nausea and/or vomiting. Cancer Treat Rev 1984; 11(3):257-264.
6. Jacobsen PB, Redd WH. The development and management of chemotherapy-related anticipatory nausea and vomiting. Cancer Invest 1988; 6(3):329-336.
7. Kris MG, Hesketh PJ, Somerfield MR et al. American Society of Clinical Oncology guideline for antiemetics in oncology: update 2006. J Clin Oncol 2006; 24(18):2932-2947.
8. Harding RK. Prodromal effects of radiation: pathways, models, and protection by antiemetics. Pharmacol Ther 1988; 39(1-3):335-345.
9. Dodds LJ. The control of cancer chemotherapy-induced nausea and vomiting. J Clin Hosp Pharm 1985; 10(2):143-166.
10. BORISON HL, WANG SC. Physiology and pharmacology of vomiting. Pharmacol Rev 1953; 5(2):193-230.
11. Grunberg SM, Osoba D, Hesketh PJ et al. Evaluation of new antiemetic agents and definition of antineoplastic agent emetogenicity–an update. Support Care Cancer 2005; 13(2):80-84.
12. Grunberg SM, Koeller JM. Palonosetron: a unique 5-HT3-receptor antagonist for the prevention of chemotherapy-induced emesis. Expert Opin Pharmacother 2003; 4(12):2297-2303.
13. Shadle CR, Lee Y, Majumdar AK et al. Evaluation of potential inductive effects of aprepitant on cytochrome P450 3A4 and 2C9 activity. J Clin Pharmacol 2004; 44(3):215-223.
PROSE: Randomized proteomic stratified phase III study of second line erlotinib versus chemotherapy in patients with inoperable non–small cell lung cancer (NSCLC)
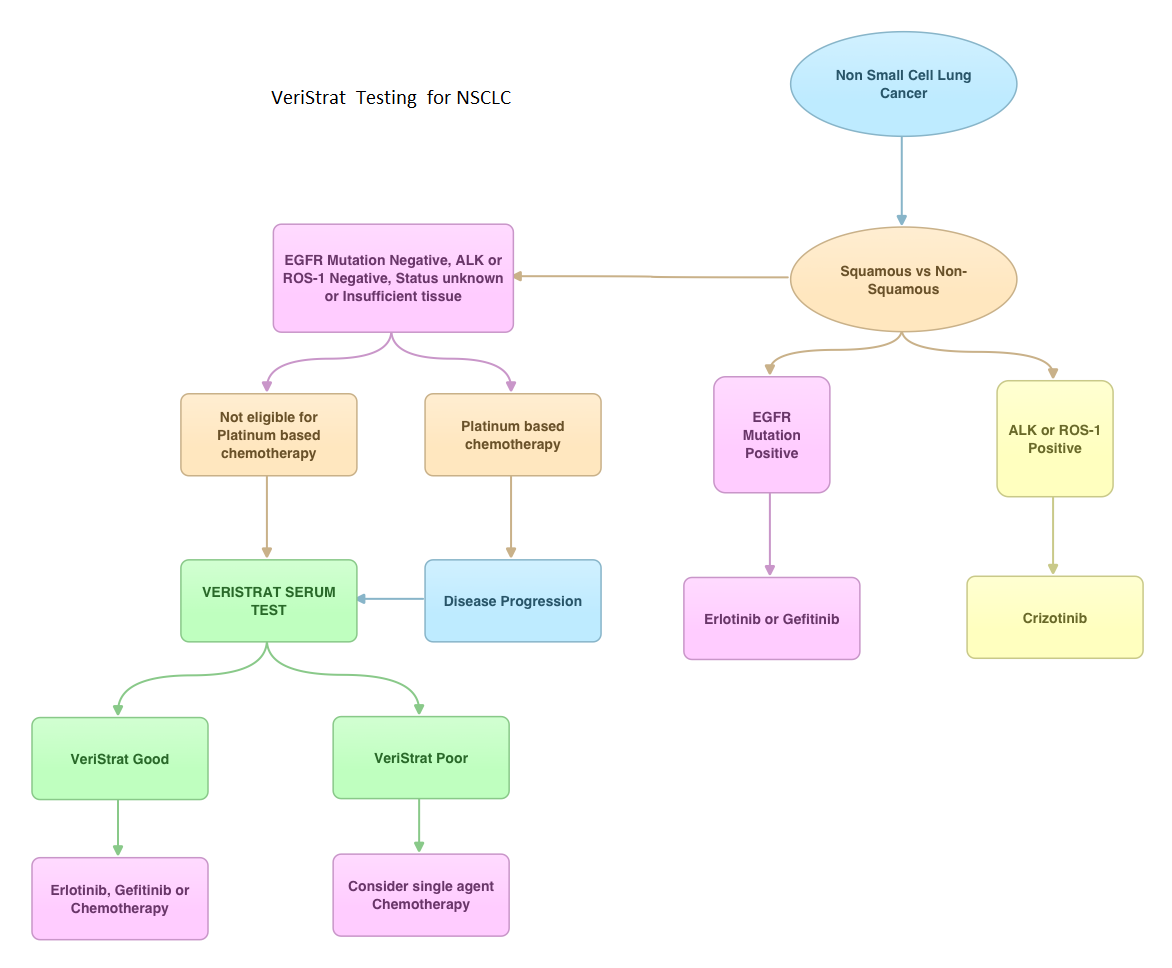
SUMMARY: VeriStrat ® is a clinically validated serum/plasma-based assay, for patients with advanced Non Small Cell Lung Cancer (NSCLC). VeriStrat® is a serum test of prognostic and predictive value that classifies patients as VeriStrat-Good (VS-G) or VeriStrat-Poor (VS-P) based on eight mass spectral peaks or proteomic patterns of the patients serum. Proteomics is the large-scale study of protein structure and functions. VeriStrat® testing is protein based and therefore has no correlation with known genomic biomarkers. It is well established that EGFR-TKIs (Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors) are more effective in NSCLC patients with EGFR activating mutations. PROSE is a multicenter, double blind, randomized, VeriStrat® stratified, phase III study. In this trial, over 90% of the patients had no EGFR mutations (EGFR-Wild Type). Two hundred and eighty five (285) patients with advanced NSCLC who had first line treatment regimen with platinum-based therapy were randomly assigned to receive second line chemotherapy (CT) with single agent ALIMTA® (Pemetrexed) or TAXOTERE® (Docetaxel), at standard doses (N=129) or TARCEVA® (Erlotinib) 150 mg po qd (N=134). Patients and study investigators were blinded to the patients VeriStrat® status. Patients were classified as VeriStrat-Good or VeriStrat-Poor based on the VeriStrat® results. Patients in the treatment groups were stratified by age, gender, tumor histology, ECOG-PS and smoking history. Crossover was permitted upon disease progression. The primary objective of the study was to demonstrate differential treatment benefit between TARCEVA® and CT with regards to Overall Survival (OS). Median overall survival (OS) was 9 months for the patients in the CT group and 7.7 months for TARCEVA® group and this was not statistically significant (P=0.3). However when evaluated by VeriStrat® status, CT was beneficial for the VeriStrat-Poor patients compared to TARCEVA®, with significantly better median OS (6.3 vs 3 months, P=0.02). Age, gender, histology (squamous vs non-squamous) and smoking history had no impact on the overall survival. The authors concluded that patients classified as VeriStrat-Poor have better survival with CT than TARCEVA®, whereas patients classified as VeriStrat-Good have similar survival with TARCEVA® and CT. VeriStrat® testing therefore, can help physicians choose between TARCEVA® and CT, for their patients with advanced NSCLC. This test helps physicians identify patients who are likely to have good or poor outcomes after treatment with EGFR inhibitors and thereby can provide valuable insight into whether CT or targeted therapy with TARCEVA®, a EGFR-TKI, is appropriate for their patients with advanced NSCLC, in the second line setting. This information is especially important for patients without an EGFR mutation or for those, whose EGFR mutation status is unknown. Sorlini C, Barni S, Petrelli F, et al. J Clin Oncol 29: 2011 (suppl; abstr TPS214)
TTP, HUS and aHUS: Different diseases – Different treatments
SUMMARY: Thrombotic Thrombocytopenic Purpura (TTP), Hemolytic Uremic Syndrome (HUS) and Atypical Hemolytic Uremic Syndrome (aHUS) are Thrombotic Microangiopathies (TMA’s) associated with MicroAngiopathic Hemolytic Anemia and thrombocytopenia. Even though their clinical presentation has some similarities, they are distinct entities with different pathophysiology and hence managed differently. With the identification of von Willebrand Factor (vWF) cleaving protease ADAMTS13 (A disintegrin and metalloprotease with thrombospondin type 1 repeats, member 13) in 1996, we are now able to better understand and appropriately manage these TMA’s. Patients with TTP are deficient in ADAMTS13 and therefore develop platelet microthrombin in small blood vessels due to uninhibited propagation of platelet aggregates bound to ultra high molecular weight VWF multimers. Approximately 10% or less of Shiga-Toxin producing Escherichia Coli (STEC) infections may be associated with HUS. aHUS is caused by a genetic deficiency of one or more complement regulatory proteins which results in uncontrolled activity of the alternate complement pathway. Plasma Exchange in TTP restores the protease activity of ADAMTS13 whereas aHUS is treated with SOLIRIS® (Eculizumab) to inhibit complement mediated TMA. Once a diagnosis of STEC-HUS is confirmed, hospitalization and intensive care with transfusions and kidney dialysis may become necessary. George JN. Blood 2010:116; 4060-4069
