
The FDA on January 16, 2025, granted traditional approval to CALQUENCE® (Acalabrutinib) with Bendamustine and Rituximab for adults with previously untreated Mantle Cell Lymphoma (MCL) who are ineligible for autologous Hematopoietic Stem Cell Transplantation (HSCT). CALQUENCE® is a product of AstraZeneca.
Tag: Non-Hodgkin Lymphoma
ZYNLONTA® in Combination with Rituximab Shows Dramatic Activity in High Risk Follicular Lymphoma
SUMMARY: The American Cancer Society estimates that in 2025, about 80,350 people will be diagnosed with Non Hodgkin Lymphoma (NHL) in the United States and about 19,390 individuals will die of this disease. Indolent Non-Hodgkin Lymphomas are mature B cell lymphoproliferative disorders and include Follicular Lymphoma, Nodal Marginal Zone Lymphoma (NMZL), Extranodal Marginal Zone Lymphoma (ENMZL) of Mucosa-Associated Lymphoid Tissue (MALT), Splenic Marginal Zone Lymphoma (SMZL), LymphoPlasmacytic Lymphoma (LPL) and Small Lymphocytic Lymphoma (SLL).
Follicular Lymphoma is the most indolent form and second most common form of all NHLs, and they are a heterogeneous group of lymphoproliferative malignancies. Approximately 20% of all NHLs are Follicular Lymphomas and the average age of diagnosis is 65 years. Advanced stage indolent NHL is not curable and as such, prolonging Progression Free Survival (PFS) and Overall Survival (OS), while maintaining Quality of Life, have been the goals of treatment intervention. Asymptomatic patients with indolent NHL are generally considered candidates for “watch and wait” approach. Patients with advanced stage symptomatic Follicular Lymphoma are often treated with induction chemoimmunotherapy followed by maintenance RITUXAN® (Rituximab). This can result in a median Progression Free Survival (PFS) of 6-8 yrs and a median Overall Survival (OS) of 12-15 yrs. However, approximately 30% of the patients will relapse in 3 years, with prognosis worsening after each subsequent relapse. Despite advances in treatment for Follicular Lymphoma, there remains an unmet need for additional options that offer treatment-free intervals with durable, complete responses.
Loncastuximab tesirine (ZYNLONTA®) is an Antibody-Drug Conjugate (ADC) and consists of a humanized monoclonal antibody (anti-CD19) linked to a cytotoxic alkylating agent (pyrrolobenzodiazepine dimer, or PBD). Upon binding to CD19 on B-calls, Loncastuximab is internalized into the cells and PBD is released inside the cells, where it crosslinks DNA and induces tumor cell death. Loncastuximab is presently indicated for the treatment of adult patients with relapsed or refractory Large B-Cell Lymphoma after two or more lines of systemic therapy, including Diffuse Large B-Cell Lymphoma (DLBCL) not otherwise specified, DLBCL arising from low-grade lymphoma, and high-grade B-cell lymphoma. Preliminary data suggested promising activity of Loncastuximab in Follicular Lymphoma, with and synergistic activity between Rtuximab-induced cytotoxicity and Loncastuximab.
The researchers in this study evaluated Loncastuximab tesirine combined with Rituximab for second-line and later treatment of Follicular Lymphoma. This single institution, investigator-initiated, Phase 2 trial enrolled 39 patients with Grade 1-3A Relapsed/Refractory Follicular Lymphoma treated with one or more lines of therapy, who presented with progression of disease within 24 months (POD24). Treatment for the initial 21 weeks consisted of Rituximab 375 mg/m2 IV weekly for 4 weeks followed by 1 dose every 8 weeks for a total of 5 doses in combination with Loncastuximab 0.15 mg/kg IV every 3 weeks for 2 doses followed by 0.075 mg/kg IV every 3 weeks for a total of 7 doses. Premedication with Dexamethasone 4 mg twice daily for 3 days was required. Patients achieving Complete Response at Week 21 discontinued Loncastuximab and received two more doses of Rituximab every 8 weeks. Patients with a partial response at week 21 continued both agents for 18 more weeks. No prophylaxis was required per study protocol. The median age of patients was 68 years and majority of patients were men (54%) with advanced-stage (82%), high-disease burden by GELF criteria (92%), and/or POD24 after frontline immunochemotherapy (51%). The median FLIPI score was 3 with most patients assigned to the high-risk group (61.5%). Median lines of prior therapy were 1, and R-CHOP was the most common first-line therapy (56.5%), followed by Bendamustine with Rituximab (25.6%), single-agent Rituximab (15.3%), and Fludarabine, Mitoxantrone, and Dexamethasone (2.6%). The Primary endpoint was Complete Metabolic Response (CMR) rate at week 12, assessed by the Lugano 2014 classification, in patients who had received at least three doses of Loncastuximab. The safety analysis included all patients who received one or more doses of Loncastuximab.
At a median follow-up of 18.2 months, the Objective Response Rate (ORR) at week 12 was 97.1%, with a CMR rate of 68.6% and a Partial Metabolic Response (PMR) rate of 28.6%. The CMR rate improved to 80% at week 21. All patients who achieved a CMR maintained their response. Baseline bone marrow involvement resolved in all patients (N=10) at week 12 reassessment. The response rates were similar in patients with POD24 and those without, each of which made up roughly half the patients in the trial. At 18 months, the Progression Free Survival (PFS) rate was 90.1% and the Overall Survival (OS) rate was 93.3%, and the median PFS and OS was not reached. The main adverse events with this therapy were skin rash that worsened with sun exposure, and fluid retention, which could be treated with diuretics.
In conclusion, a combination of Loncastuximab with Rituximab demonstrated dramatic activity with robust Complete Metabolic Rate and promising survival benefit with manageable toxicities, in patients with high risk Relapsed or Refractory Follicular Lymphoma. The results of this study support this combination as a new treatment option for this patient group, and multicenter expansion cohort is ongoing. Because of the high Complete Response by week 12, the researchers recently reduced the treatment length from 10 to 6 months, with the hope that relatively short course of treatment combined with lower toxicity will allow patients to better tolerate and complete the therapy.
Loncastuximab tesirine with rituximab in patients with relapsed or refractory follicular lymphoma: a single-centre, single-arm, phase 2 trial. Alderuccio JP, Alencar AJ, Schatz JH, et al. The Lancet. 2025;12:E23-E34.
FDA Approves ADCETRIS® with Lenalidomide and Rituximab for Relapsed and Refractory Diffuse Large B-Cell Lymphoma
SUMMARY: The FDA on February 11, 2025, approved Brentuximab vedotin (ADCETRIS®) in combination with Lenalidomide (REVLIMID®) and a Rituximab product for adult patients with Relapsed or Refractory Large B-Cell Lymphoma (LBCL), including Diffuse Large B-Cell Lymphoma (DLBCL) not otherwise specified (NOS), DLBCL arising from indolent lymphoma, or High-Grade B-Cell Lymphoma (HGBL), after two or more lines of systemic therapy who are ineligible for Autologous hematopoietic Stem Cell Transplantation (auto-HSCT) or CAR T-cell therapy.
The American Cancer Society estimates that in 2025, about 80,350 people will be diagnosed with Non Hodgkin Lymphoma (NHL) in the United States and about 19,390 individuals will die of this disease. Diffuse Large B-Cell Lymphoma (DLBCL) is the most common of the aggressive Non-Hodgkin lymphomas in the United States, and more than 25,000 cases of DLBCL are diagnosed each year in the United States, accounting for more than 25 percent of all lymphoma cases. The incidence has steadily increased 3-4% each year. More than half of patients are 65 or older at the time of diagnosis and the incidence is likely to increase with aging of the American population. The etiology of Diffuse Large B-Cell Lymphoma is unknown. Contributing risk factors include immunosuppression (AIDS, transplantation setting, autoimmune diseases), UltraViolet radiation, pesticides, hair dyes, and diet. DLBCL can develop spontaneously or as a result of Richters transformation of low grade diseases such as Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma, Follicular Lymphoma, or Marginal Zone Lymphoma.
DLBCL is a neoplasm of large B cells and the most common chromosome abnormality involves alterations of the BCL-6 gene at the 3q27 locus, which is critical for germinal center formation. Two major molecular subtypes of DLBCL arising from different genetic mechanisms have been identified, using Gene Expression Profiling: Germinal Center B-cell-like (GCB) and Activated B-Cell-like (ABC). Patients in the GCB subgroup have a higher 5-year survival rate, independent of clinical IPI (International Prognostic Index) risk score, whereas patients in the ABC subgroup have a significantly worse outcome. Regardless of molecular subtype, R-CHOP regimen (Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone), given every 21 days, for 6 cycles, delivered with curative intent, is the current standard of care for patients of all ages, with newly diagnosed DLBCL. Approximately 30-40% of patients experience disease progression or relapse during the first 2 years and attempts to improve on R-CHOP regimen have not been successful. Maintenance treatment strategy following R-CHOP, to better control the disease, delay disease progression and improve long term survival, have included Autologous Stem Cell Transplantation, CAR T-cell therapy, maintenance treatment with agents such as oral protein kinase inhibitor Enzastaurin and Everolimus. Outcomes for transplant-ineligible patients with Relapsed/Refractory DLBCL patients remain poor. There is a critical unmet need for this patient group.
Brentuximab Vedotin (ADCETRIS®) is an Antibody-Drug Conjugate (ADC) that targets CD30, which is a surface antigen, expressed on Reed-Sternberg cells, in patients with Classical Hodgkin lymphoma. This ADC consists of an anti-CD30 monoclonal antibody linked to MonoMethyl Auristatin E (MMAE), an antimicrotubule agent. Upon binding to the CD30 molecule on the cancer cells, MMAE is released into the cancer cell, resulting in cell death. Preclinical data and early phase studies provided the rationale for combining Brentuximab vedotin, Lenalidomide and Rituximab for the treatment of Relapsed/Refractory DLBCL.
ECHELON-3 is an ongoing, global, randomized, double-blind, multicenter Phase III study, designed to evaluate the efficacy and safety of Brentuximab vedotin in combination with Lenalidomide and Rituximab, compared to Lenalidomide and Rituximab plus placebo, in adult patients with Relapsed/Refractory DLBCL, regardless of CD30 expression, who have received two or more prior lines of therapy and were ineligible for or had previously failed Hematopoietic Stem Cell Transplant (HSCT) or Chimeric Antigen Receptor (CAR) T-cell therapy.
In this global study, 230 patients were randomized 1:1 to receive either Brentuximab vedotin plus Lenalidomide and Rituximab (BV+R2) – N=112, with Brentuximab vedotin administered at 1.2 mg/kg IV every 3 weeks, Lenalidomide at 20 mg orally daily, and Rituximab at 375 mg/m² IV every 3 weeks, or Placebo plus Lenalidomide and Rituximab (placebo+R2) – N=118. Placebo for Brentuximab vedotin with Lenalidomide and Rituximab was administered in the same manner. Treatment continued until disease progression or unacceptable toxicity. The median age was 71 years, 56% were male, median prior lines of therapy was 3, 29% had prior CAR T-cell therapy, approximately 15% of patients had prior bispecific antibody exposure, and 68% were CD30 negative with CD30 tumor expression of less than 1%. The Primary endpoint was Overall Survival (OS). Secondary endpoints included Progression-Free Survival (PFS), Objective Response Rate (ORR) and Complete Response (CR) Rate.
At the interim analysis, with a median follow-up of 16.4 months, BV+R2 regimen demonstrated a median OS of 13.8 months compared to 8.5 months with placebo+R2, representing a 37% reduction in the risk of death (HR=0.63; P=0.009). The median PFS was 4.2 months in the BV+R2 arm versus 2.6 months in the placebo+R2 arm (HR=0.53; P<0.001). The ORR was 64% with BV+R2 compared to 42% with placebo+R2 (P=0.001). The CR rate was 40% with BV+R2 versus 19% with placebo+R2. The efficacy benefits of BV+R2 were consistent across key subgroups, including patients with CD30-positive and CD30-negative disease, highlighting the broad applicability of the regimen.
Grade 3 or higher adverse events were more frequent in the BV+R2 arm compared to placebo+R2 (88% versus 77%), and the most common Grade 3 or higher adverse events included neutropenia, anemia and diarrhea. Peripheral Neuropathy was higher with BV+R2 compared to placebo+R2, although generally manageable.
In conclusion, the ECHELON-3 study demonstrated that the addition of Brentuximab vedotin to Lenalidomide and Rituximab significantly improved Overall Survival, Progression-Free Survival, and Response Rates in patients with Relapsed/Refractory DLBCL, compared to Lenalidomide and Rituximab alone. This regimen offers a promising new treatment option for patients who have exhausted standard therapies or are ineligible for intensive treatments like CAR-T cell therapy or HSCT. The results underscore the potential of targeted therapies in reshaping the management of DLBCL, providing renewed hope for improved outcomes in this challenging disease setting. As the study continues to follow patients for long-term outcomes, ongoing research will further elucidate the durability of responses and additional safety data, thereby informing future clinical practice guidelines and optimizing patient care strategies.
Brentuximab Vedotin Combination for Relapsed Diffuse Large B-Cell Lymphoma. Bartlett NL, Hahn U, Kim W-S, et al. J Clin Oncol. January 07,2025. https://doi.org/10.1200/JCO-24-02242.
EPKINLY® (Epcoritamab-bysp)
The FDA on June 26, 2024, granted accelerated approval to EPKINLY®, a bispecific CD20-directed CD3 T-cell engager, for adult patients with relapsed or refractory Follicular Lymphoma (FL) after two or more lines of systemic therapy. EPKINLY® is a product of Genmab US, Inc.
BREYANZI® (Lisocabtagene maraleucel)
The FDA on May 30, 2024, approved BREYANZI® for adult patients with relapsed or refractory Mantle Cell Lymphoma (MCL) who have received at least two prior lines of systemic therapy, including a Bruton Tyrosine Kinase Inhibitor (BTKi). BREYANZI® is a product of Juno Therapeutics, Inc.
BREYANZI® (Lisocabtagene maraleucel)
The FDA on May 15, 2024, granted accelerated approval to BREYANZI® for adults with relapsed or refractory Follicular Lymphoma (FL) who have received two or more prior lines of systemic therapy. BREYANZI® is a product of Juno Therapeutics, Inc.
FDA Approves LYMPHIR® for Relapsed and Refractory Cutaneous T-Cell Lymphoma
SUMMARY: The FDA on August 7, 2024 approved LYMPHIR® (Denileukin diftitox-cxdl), a novel immunotherapy for the treatment of relaped/refractory Cutaneous T-Cell Lymphoma (CTCL) after at least one prior systemic therapy.
Primary Cutaneous T Cell Lymphoma is a type of Non Hodgkin Lymphoma and includes a spectrum of diseases that primarily involve the skin, but may ultimately involve lymph nodes, blood and visceral organs such as spleen, liver and lungs. CTCL initially presents as red, scaly, itchy patches on the skin and is often misdiagnosed as eczema. Approximately 3,000 new cases are reported in the United States every year, with an estimated 30,000 – 40,000 individuals living with the disease. The incidence is higher in blacks than Caucasians or Asians. It is more common in men, and is typically first diagnosed in patients between the ages of 50 and 60 years of age. Mycosis Fungoides/Sezary Syndrome is the most common type of CTCL. There is currently no curative therapy for advanced CTCL other than Allogeneic Stem Cell Transplantation. Patients often receive several skin-directed therapies as well as systemic therapies to achieve effective disease control, but the disease eventually becomes refractory. There is therefore an unmet clinical need for novel therapies.
Denileukin diftitox is a recombinant fusion protein composed of Interleukin-2 (IL-2) receptor binding domain and diphtheria toxin fragments. It selectively targets IL-2 receptor-expressing cells, causing diphtheria toxin fragments that have entered cells to inhibit protein synthesis, leading to apoptosis. Its unique mechanism of action targets both malignant T-cells and immunosuppressive regulatory T-cells (Tregs). Transiently eliminating Tregs has the potential of unleashing potent immune responses by the immune system of the patient against their tumors. This agent was approved and marketed as ONTAK® in the US from 1999-2014 for the treatment of relapsed/refractory CTCL. However, the product was withdrawn from the market in 2014 because of manufacturing issues related to its bacterial expression. Manufacturing improvements to decrease the presence of misfolded and aggregated proteins resulted in a new and more purified bioactive formulation LYMPHIR®, that has 1.5-2 times greater specific bioactivity in non-clinical assays compared with ONTAK®. It is considered a new drug by the FDA, requiring a new registrational clinical trial.
The efficacy and safety of LYMPHIR® was assessed in a multicenter, open-label, Phase III Pivotal Study 302 of CTCL patients, who had previously received at least one systemic treatment. This analysis included 69 patients (N=69) with Stage I-III CTCL who had CD25 expression on at least 20% of biopsied malignant cells per immunohistochemistry and had received a median four prior therapies. The median age was 64 years, 65% were men, 73% were White and 19% were African American, 66 patients had Mycosis fungoides and 3 had Sezary syndrome. Approximately 30% had Stage I disease, 48% had Stage II disease and 22% had Stage III disease. Patients received LYMPHIR® 9 mcg/kg/day for 5 days, every 21 days for up to 8 cycles. The Primary efficacy outcome measure was Objective Response Rate (ORR), as assessed by an Independent Review Committee (IRC).
The Objective Response Rate was 36.2%, with 8.7% achieving a Complete Response. The median time to response was 1.41 months, with 70% of responders seeing results after 1-2 cycles of treatment. Duration of response was at least 6 months for 52% of the patients and 84% (54/64) of skin evaluable patients had a decrease in skin tumor burden, and 12.5% (8/64) saw complete clearing of skin disease. Pruritis was evaluated as an exploratory endpoint with 31.7% of patients demonstrating clinically significant pruritus improvement. Importantly, no cumulative toxicity was observed in patients receiving LYMPHIR®.
The safety profile of LYMPHIR® was consistent with the known safety profile for Denileukin diftitox. Across three studies of 119 CTCL patients receiving 9 μg dose of Denileukin diftitox, the most common (20% or more) adverse reactions including laboratory abnormalities were, infusion reactions, increased transaminases, decreased albumin, nausea, edema, anemia, fatigue, musculoskeletal pain, rash, chills, constipation, pyrexia, blurred vision and Capillary Leak Syndrome (CLS). Infusion reactions, CLS, and visual impairment were mostly Grade 1/2 and effectively managed.
In conclusion, LYMPHIR® represents a significant advancement in treating Cutaneous T-Cell Lymphoma, especially for patients who have not responded well to previous treatments. As the first systemic option in years targeting the IL-2 receptor on malignant T-cells and Tregs, LYMPHIR® provides new hope for managing the long-term challenges of CTCL, such as severe skin symptoms and secondary infections. This development moves us closer to addressing the needs of CTCL patients who struggle with ongoing disease despite prior therapies.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/761312s000lbl.pdf
What is the Real-World Evidence for the Effectiveness of Mogamulizumab in Patients with Mycosis Fungoides and Sézary Syndrome?
Written by: Francine Foss, MD
Professor of Medicine (Hematology) and Dermatology
Director, Multidisciplinary T cell Lymphoma Program, Hematology; Scientific Leader, Lymphoma CRT
Yale Cancer Center
Content sponsored by Kyowa Kirin, Inc.
Dr. Foss is a paid consultant for Kyowa Kirin and was compensated for her contribution in drafting this article.
POTELIGEO® (mogamulizumab–kpkc), indicated for the treatment of adult patients with relapsed or refractory Mycosis Fungoides (MF) or Sézary Syndrome (SS) after at least one prior systemic therapy, is a first-in-class humanized monoclonal antibody (mAb) directed against CC chemokine receptor 4 (CCR4), a protein consistently expressed on cancerous cells seen in both MF and SS.1-3
POTELIGEO received FDA approval based on results of the MAVORIC trial, a randomized, open-label, phase 3 trial that compared its efficacy with that of an active comparator, vorinostat, in previously treated patients with relapsed or refractory MF or SS. In MAVORIC, patients receiving mogamulizumab (n=186) demonstrated efficacy superior to those receiving vorinostat (n=186) in the prespecified primary endpoint, with significantly longer progression-free survival (PFS) (7.6 vs 3.1 months; hazard ratio: 0.53, 95% CI [0.41, 0.69], P<0.001). For the secondary endpoint, overall response rate (ORR), significantly more patients achieved a response to mogamulizumab vs the comparator (28% vs 5%, P<0.001). When evaluated by disease compartment, response rates were higher with mogamulizumab compared with vorinostat in the blood (67% vs 18%), skin (42% vs 16%), and lymph nodes (15% vs 4%). The most common adverse reactions (reported in ≥20% of patients) were rash, infusion-related reactions, fatigue, diarrhea, musculoskeletal pain, and upper respiratory tract infection.4
The OMEGA study was a retrospective analysis of real-world patients receiving mogamulizumab in 14 centers throughout France. The full study-report article can be accessed at https://doi.org/10.1111/jdv.19134. A total of 122 patients were reviewed, 53 with MF and 69 with SS. All had been treated with mogamulizumab from February 2014 until March 2020.5 The OMEGA study contains information that is not included in the FDA-approved labeling for POTELIGEO; the study included 2 patients with SS in whom mogamulizumab was administered as a first-line therapy. OMEGA also reported a serious adverse reaction, vitiligo (3 patients [2.4%]), that was not captured in the MAVORIC trial. It is not known if there were variations from the FDA-approved labeling in the dosing schedule for any patients included in the study. Also, OMEGA differed from MAVORIC by defining treatment responses as complete or partial response (CR or PR) that occurred at any time, and for no prespecified duration post-initiation of mogamulizumab. In MAVORIC, by definition, treatment responses were required to be confirmed CR or PR at 2 or more consecutive assessments spaced at least 8 weeks apart.5
In the OMEGA study, key outcome measures were:
Primary endpoint: Best overall response rate (bORR)a
Secondary endpoints: bORR by compartment (skin, blood, lymph nodes, viscera)b and safety
Exploratory endpoint: PFS
a Percentage of patients achieving a global overall response (CR or PR) at any time and for no prespecified duration.
b Percentage of patients achieving a CR or PR in the specified compartment at any time and for no prespecified duration.
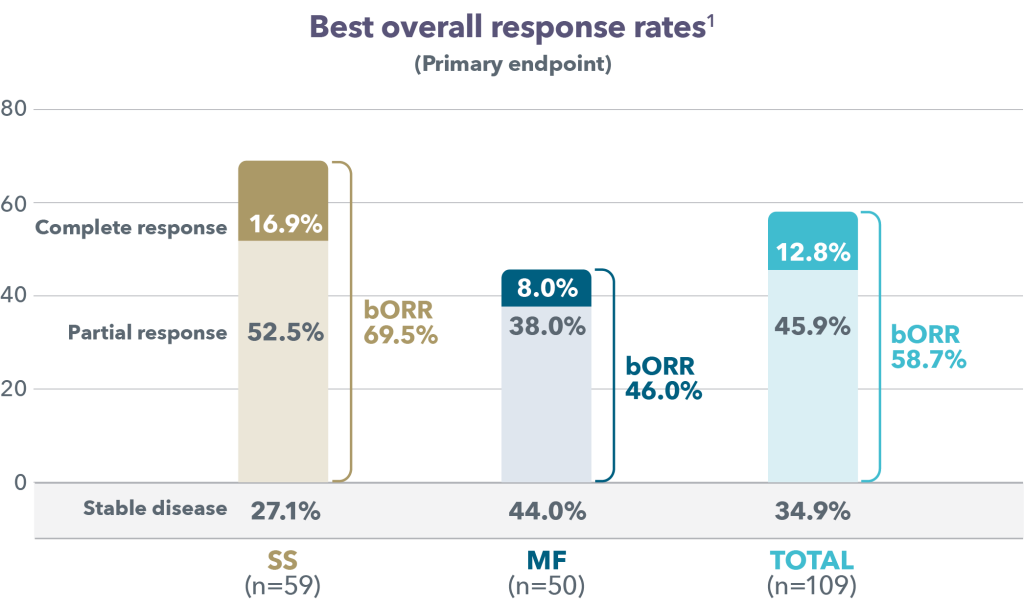
As shown in Figure 1, in the entire patient population, bORR was 58.7% (12.8% CR; 45.9% PR). In patients with SS, bORR was 69.5% (16.9% CR; 52.5% PR), and in patients with MF, bORR was 46.0% (8.0% CR; 38.0% PR). The median time to response under treatment (CR or PR) was similar according to disease subtype (3.1 months for patients with SS and MF; respective ranges: 01–25.0, and 0.3–44.3 months).5
Responses were seen across all involved disease compartments, as seen in Table 1.5

As seen in Figure 2, in the overall analysis population (n=122), the median PFS was estimated at 15.0 months (95% CI [9.0–50.8]). It was longer in patients with SS than in patients with MF (20.3 months [11.7–not reached] vs. 8.8 months [4.6–43.0]) but no significant difference between disease subtypes was shown (P= 0.0542).5

The percentage of patients who experienced a serious adverse reaction (AR) was consistent with MAVORIC (18.5% vs 20% in MAVORIC).1,2 Discontinuations of mogamulizumab due to ARs occurred in 15 patients (12.1%) rash was the most common reason for permanent discontinuation (9 patients; 7.3%). One patient discontinued due to thrombopenia and one patient discontinued due to an infusion-related reaction (0.8% each).5 The most common ARs can be seen in Table 2.

Limitations of the OMEGA study include its non-interventional and retrospective design, a patient population less selective than those in clinical trials, and overlap of 20 patients that were also part of the MAVORIC trial. The results are in line with efficacy and safety data demonstrated in the global clinical trial MAVORIC, and supports the effectiveness of mogamulizumab in real-world clinical practices.
INDICATION
POTELIGEO injection for intravenous infusion is indicated for the treatment of adult patients with relapsed or refractory mycosis fungoides (MF) or Sézary syndrome (SS) after at least one prior systemic therapy.
IMPORTANT SAFETY INFORMATION
Warnings and Precautions
• Dermatologic toxicity: Monitor patients for rash throughout the course of treatment. For patients who experienced dermatologic toxicity in Trial 1, the median time to onset was 15 weeks, with 25% of cases occurring after 31 weeks. Interrupt POTELIGEO for moderate or severe rash (Grades 2 or 3). Permanently discontinue POTELIGEO for life-threatening (Grade 4) rash or for any Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).
• Infusion reactions: Most infusion reactions occur during or shortly after the first infusion. Infusion reactions can also occur with subsequent infusions. Monitor patients closely for signs and symptoms of infusion reactions and interrupt the infusion for any grade reaction and treat promptly. Permanently discontinue POTELIGEO for any life-threatening (Grade 4) infusion reaction.
• Infections: Monitor patients for signs and symptoms of infection and treat promptly.
• Autoimmune complications: Interrupt or permanently discontinue POTELIGEO as appropriate for suspected immune-mediated adverse reactions. Consider the benefit/risk of POTELIGEO in patients with a history of autoimmune disease.
• Complications of allogeneic HSCT after POTELIGEO: Increased risks of transplant complications have been reported in patients who received allogeneic HSCT after POTELIGEO. Follow patients closely for early evidence of transplant-related complications
Adverse Reactions
• The most common adverse reactions (reported in ≥10% of patients) with POTELIGEO in the clinical trial were rash, including drug eruption (35%), infusion reaction (33%), fatigue (31%), diarrhea (28%), drug eruption (24%), upper respiratory tract infection (22%), musculoskeletal pain (22%), skin infection (19%), pyrexia (17%), edema (16%), nausea (16%), headache (14%), thrombocytopenia (14%), constipation (13%), anemia (12%), mucositis (12%), cough (11%), and hypertension (10%).
Please see the full Prescribing Information for POTELIGEO at www.poteligeohcp.com for additional information.
You are encouraged to report suspected adverse reactions to Kyowa Kirin, Inc. at 1-844-768-3544 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
References:
1. Ferenczi K, et al. Increased CCR4 expression in cutaneous T cell lymphoma. J Invest Dermatol. 2002;119(6):1405-1410.
2. Yoshie O, et al. Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood. 2002;99(5):1505-1511.
3. Ishida T, et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res. 2003;9(10 Pt 1):3625-3634.
4. POTELIGEO [package insert]. Kyowa Kirin Inc., Princeton, NJ USA.
5. Beylot-Barry M, Quereux G, Nardin C, et al. Effectiveness of mogamulizumab in patients with mycosis fungoides or Sézary syndrome: a multicentre, retrospective, real-world French study. J Eur Acad Dermatol Venereol. 2023;37(9):1777-1784.
POTELIGEO is a registered trademark of Kyowa Kirin Co., Ltd.
© 2024 Kyowa Kirin, Inc. All rights reserved.
510 Carnegie Center Dr. Princeton, NJ 08540 USA
COMM-US-POT-0274 May 2024
Late Breaking Abstract – ASCO 2024: ADCETRIS® Combination Improves Overall Survival in Relapsed and Refractory Diffuse Large B-Cell Lymphoma
SUMMARY: The American Cancer Society estimates that in 2024, about 80,620 people will be diagnosed with Non Hodgkin Lymphoma (NHL) in the United States and about 20,140 individuals will die of this disease. Diffuse Large B-Cell Lymphoma (DLBCL) is the most common of the aggressive Non-Hodgkin lymphomas in the United States, and more than 25,000 cases of DLBCL are diagnosed each year in the United States, accounting for more than 25 percent of all lymphoma cases. The incidence has steadily increased 3-4% each year. More than half of patients are 65 or older at the time of diagnosis and the incidence is likely to increase with aging of the American population. The etiology of Diffuse Large B-Cell Lymphoma is unknown. Contributing risk factors include immunosuppression (AIDS, transplantation setting, autoimmune diseases), UltraViolet radiation, pesticides, hair dyes, and diet. DLBCL can develop spontaneously or as a result of Richters transformation of low grade diseases such as Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma, Follicular Lymphoma, or Marginal Zone Lymphoma.
DLBCL is a neoplasm of large B cells and the most common chromosome abnormality involves alterations of the BCL-6 gene at the 3q27 locus, which is critical for germinal center formation. Two major molecular subtypes of DLBCL arising from different genetic mechanisms have been identified, using Gene Expression Profiling: Germinal Center B-cell-like (GCB) and Activated B-Cell-like (ABC). Patients in the GCB subgroup have a higher 5-year survival rate, independent of clinical IPI (International Prognostic Index) risk score, whereas patients in the ABC subgroup have a significantly worse outcome. Regardless of molecular subtype, R-CHOP regimen (Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone), given every 21 days, for 6 cycles, delivered with curative intent, is the current standard of care for patients of all ages, with newly diagnosed DLBCL. Approximately 30-40% of patients experience disease progression or relapse during the first 2 years, and attempts to improve on R-CHOP regimen have not been successful. Maintenance treatment strategy following R-CHOP, to better control the disease, delay disease progression and improve long term survival, have included Autologous Stem Cell Transplantation, CAR T-cell therapy, maintenance treatment with agents such as oral protein kinase inhibitor Enzastaurin and Everolimus. Outcomes for transplant-ineligible patients with Relapsed/Refractory DLBCL patients remain poor. There is a critical unmet need for this patient group.
Brentuximab Vedotin (ADCETRIS®) is an Antibody-Drug Conjugate (ADC) that targets CD30, which is a surface antigen, expressed on Reed-Sternberg cells, in patients with Classical Hodgkin lymphoma. This ADC consists of an anti-CD30 monoclonal antibody linked to MonoMethyl Auristatin E (MMAE), an antimicrotubule agent. Upon binding to the CD30 molecule on the cancer cells, MMAE is released into the cancer cell, resulting in cell death. Preclinical data and early phase studies provided the rationale for combining Brentuximab vedotin, Lenalidomide and Rituximab for the treatment of Relapsed/Refractory DLBCL.
ECHELON-3 is an ongoing, global, randomized, double-blind, multicenter Phase III study, designed to evaluate the efficacy and safety of Brentuximab vedotin in combination with Lenalidomide and Rituximab, compared to Lenalidomide and Rituximab plus placebo, in adult patients with Relapsed/Refractory DLBCL, regardless of CD30 expression, who have received two or more prior lines of therapy and were ineligible for or had previously failed Hematopoietic Stem Cell Transplant (HSCT) or Chimeric Antigen Receptor (CAR) T-cell therapy.
In this global study, 230 patients were randomized 1:1 to receive either Brentuximab vedotin plus Lenalidomide and Rituximab (BV+R2) – N=112, with Brentuximab vedotin administered at 1.2 mg/kg IV every 3 weeks, Lenalidomide at 20 mg orally daily, and Rituximab at 375 mg/m² IV every 3 weeks, or Placebo plus Lenalidomide and Rituximab (placebo+R2) – N=118. Placebo for Brentuximab vedotin with Lenalidomide and Rituximab was administered in the same manner. Treatment continued until disease progression or unacceptable toxicity. The median age was 71 years, 56% were male, median prior lines of therapy was 3, 29% had prior CAR T-cell therapy, approximately 15% of patients had prior bispecific antibody exposure, and 68% were CD30 negative with CD30 tumor expression of less than 1%. The Primary endpoint was Overall Survival (OS). Secondary endpoints included Progression-Free Survival (PFS), Objective Response Rate (ORR) and Complete Response (CR) Rate.
At the interim analysis, with a median follow-up of 16.4 months, BV+R2 regimen demonstrated a median OS of 13.8 months compared to 8.5 months with placebo+R2, representing a 37% reduction in the risk of death (HR=0.63; P=0.0085). The median PFS was 4.2 months in the BV+R2 arm versus 2.6 months in the placebo+R2 arm (HR=0.53; P<0.0001). The ORR was 64.3% with BV+R2 compared to 41.5% with placebo+R2 (P=0.0006). The CR rate was 40.2% with BV+R2 versus 18.6% with placebo+R2. The efficacy benefits of BV+R2 were consistent across key subgroups, including patients with CD30-positive and CD30-negative disease, highlighting the broad applicability of the regimen.
Grade 3 or higher adverse events were more frequent in the BV+R2 arm compared to placebo+R2 (88% versus 77%), and the most common Grade 3 or higher adverse events included neutropenia, anemia and diarrhea. Peripheral Neuropathy was higher with BV+R2 compared to placebo+R2, although generally manageable.
In conclusion, the ECHELON-3 study demonstrated that the addition of Brentuximab vedotin to Lenalidomide and Rituximab significantly improved Overall Survival, Progression-Free Survival, and Response Rates in patients with Relapsed/Refractory DLBCL, compared to Lenalidomide and Rituximab alone. This regimen offers a promising new treatment option for patients who have exhausted standard therapies, or are ineligible for intensive treatments like CAR-T cell therapy or HSCT. The results underscore the potential of targeted therapies in reshaping the management of DLBCL, providing renewed hope for improved outcomes in this challenging disease setting. As the study continues to follow patients for long-term outcomes, ongoing research will further elucidate the durability of responses and additional safety data, thereby informing future clinical practice guidelines and optimizing patient care strategies.
Brentuximab vedotin in combination with lenalidomide and rituximab in patients with relapsed/refractory diffuse large B-cell lymphoma: Results from the phase 3 ECHELON-3 study. Kim JA, Hahn U, Kim W-S, et al. Journal of Clinical Oncology. Volume 42, Number 17_suppl. https://doi.org/10.1200/JCO.2024.42.17_suppl.LBA7005
FDA Approves BREYANZI® for Relapsed/Refractory Follicular Lymphoma
SUMMARY: The FDA on May 15, 2024, granted accelerated approval to BREYANZI® (Lisocabtagene maraleucel) for adults with Relapsed or Refractory Follicular Lymphoma who have received two or more prior lines of systemic therapy. The American Cancer Society estimates that in 2024, about 80,620 people will be diagnosed with Non-Hodgkin Lymphoma (NHL) in the United States and about 20,140 individuals will die of this disease. Indolent Non-Hodgkin Lymphomas are mature B cell lymphoproliferative disorders and include Follicular Lymphoma, Nodal Marginal Zone Lymphoma (NMZL), Extranodal Marginal Zone Lymphoma (ENMZL) of Mucosa-Associated Lymphoid Tissue (MALT), Splenic Marginal Zone Lymphoma (SMZL), LymphoPlasmacytic Lymphoma (LPL) and Small Lymphocytic Lymphoma (SLL).
Follicular Lymphoma is the most indolent form and second most common form of all NHLs and they are a heterogeneous group of lymphoproliferative malignancies. Approximately 20% of all NHLs are Follicular Lymphomas and the average age of diagnosis is 65 years. Advanced stage indolent NHL is not curable and as such, prolonging Progression Free Survival (PFS) and Overall Survival (OS), while maintaining Quality of Life, have been the goals of treatment intervention. Asymptomatic patients with indolent NHL are generally considered candidates for “watch and wait” approach. Patients with advanced stage symptomatic Follicular Lymphoma are often treated with induction chemoimmunotherapy followed by maintenance RITUXAN® (Rituximab). This can result in a median Progression Free Survival (PFS) of 6-8 yrs and a median Overall Survival (OS) of 12-15 yrs. However, approximately 30% of the patients will relapse in 3 years, with prognosis worsening after each subsequent relapse. Despite advances in treatment for Follicular Lymphoma, there remains an unmet need for additional options that offer treatment-free intervals with durable, complete responses.
Chimeric Antigen Receptor (CAR) T-cell therapy is a type of immunotherapy and consists of T cells collected from the patient’s blood in a leukapheresis procedure, and genetically engineered to produce special receptors on their surface called Chimeric Antigen Receptors (CAR). These reprogrammed cytotoxic T cells with the Chimeric Antigen Receptors on their surface are now able to recognize a specific antigen on tumor cells. These genetically engineered and reprogrammed CAR T-cells are grown in the lab and are then infused into the patient. These cells in turn proliferate in the body of patients and the engineered receptor on the cell surface help recognize and kill cancer cells that expresses that specific antigen.
Patients, following treatment with CAR T-cells, develop B-cell aplasia (absence of CD19 positive cells) due to B-cell destruction and may need immunoglobin replacement. Hence, B-cell aplasia can be a useful therapeutic marker, as continued B-cell aplasia has been seen in all patients who had sustained remission, following CAR T-cell therapy. Cytokine Release Syndrome, an inflammatory process, is the most common and serious side effect of CAR T-cell therapy and is associated with marked elevation of Interleukin-6. Cytokine release is important for T-cell activation and can result in high fevers and myalgias. This is usually self limiting although if severe can be associated with hypotension and respiratory insufficiency. Tocilizumab (ACTEMRA®), an Interleukin-6 receptor blocking antibody, produces a rapid improvement in symptoms. This is however not recommended unless the symptoms are severe and life threatening, as blunting the cytokine response can in turn negate T-cell proliferation. Elevated serum ferritin and C-reactive protein levels are surrogate markers for severe Cytokine Release Syndrome. The CAR T-cells have been shown to also access sanctuary sites such as the CNS and eradicate cancer cells. CD19 antigen is expressed by majority of the B-cell malignancies and therefore most studies using CAR T-cell therapy have focused on the treatment of advanced B-cell malignancies.
BREYANZI® is a CD19-directed genetically modified autologous T cell immunotherapy, that seeks out cancer cells expressing the antigen CD19, which is found uniquely on B cells and destroy them. BREYANZI® contains a 4-1BB costimulatory domain, which enhances the expansion and persistence of the CAR T cells. BREYANZI® was previously approved in the US for the treatment of Relapsed or Refractory Large B-Cell Lymphoma (LBCL) after at least one prior line of therapy, and also received accelerated approval for the treatment of Relapsed or Refractory Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma after at least two prior lines of therapy.
TRANSCEND-FL is a global, multicenter, open-label, single-arm Phase II trial which included patients with Relapsed or Refractory Follicular Lymphoma after two or more lines of systemic therapy including an anti-CD20 antibody and an alkylating agent. This study included 94 eligible patients (N=94) and these patients also needed to have an ECOG PS of 1 or less, as well as adequate bone marrow function to receive lymphodepleting chemotherapy. Eligible patients had PET-positive disease at baseline or after bridging therapy and had at least 9 months of follow up from first response. Patients were excluded if they had evidence or a history of composite Diffuse Large B-Cell Lymphoma and Follicular Lymphoma, or transformed Follicular Lymphoma, a WHO subclassification of duodenal-type Follicular Lymphoma, CNS-only involvement by malignancy, or prior CAR T-cell therapy or other genetically modified cell therapy. Following apheresis and collection of T cells, patients received lymphodepleting chemotherapy consisting of Fludarabine 30 mg/m2 IV and Cyclophosphamide 300 mg/m2 IV daily for 3 days. Patients could receive bridging therapy for disease control following apheresis and prior to lymphodepletion and subsequent administration of BREYANZI®. Patients received a single dose of BREYANZI® 2-7 days, following the completion of lymphodepleting chemotherapy at a target dose of 100 x 106 CAR-positive T cells.
The Primary efficacy endpoint was the Overall Response Rate (ORR), defined as the percentage of patients achieving a Partial or Complete Response per Lugano criteria as assessed by an Independent Review Committee (IRC). Secondary endpoints included Complete Response (CR) rate, Duration of Response (DOR), Progression-Free Survival (PFS), Overall Survival (OS), and Safety. Responses were evaluated using PET scans, providing a comprehensive assessment of treatment efficacy.
The study demonstrated impressive efficacy outcomes, with an ORR of 95.7% and a CR rate of 73.4% in the primary analysis set. Responses were rapid, with a median time to response of one month. After a median follow up of 16.8 months, the median Duration of Response was Not Reached. Approximately 81% of responders remained in response at 12 months, and 77% of responders remained in response at 18 months, underscoring the potential of this therapy to induce long-lasting remissions. The most common non-laboratory adverse reactions were Cytokine Release Syndrome (CRS), headache, musculoskeletal pain, fatigue, constipation, and fever.
In this largest primary analysis assessing CAR T-cell therapy for Relapsed or Refractory Follicular Lymphoma, a treatment option with a one-time infusion of BREYANZI® with the potential for lasting remission, addresses the unmet need of these patients, heralding a new era in the management of this challenging disease.
https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-lisocabtagene-maraleucel-follicular-lymphoma