Written by: Benjamin L. Kitchens, MD
Sponsored by: Jazz Pharmaceuticals
Gastroesophageal adenocarcinoma (GEA) includes cancers of the gastroesophageal junction (GEJ) and stomach. In 2026, U.S. estimates project 31,510 new cases of gastric cancer and 22,530 new cases of esophageal cancer [1,2]. Gastric and GEJ cancer are often diagnosed at advanced stages due to lack of screening programs in the US; this portends a poor prognosis. The 5-year relative survival for gastric cancer is 78.1% for localized disease, 39.0% for regional disease, and 8.1% for distant metastatic disease [1]. Similarly, esophageal cancer 5-year relative survival rates are 48.6% for localized disease, 29.1% for regional disease, and 5.3% for distant metastatic disease [2]. The poor prognoses for patients with metastatic GEA underscore the need for continued advances in early detection and innovative therapies to improve outcomes.
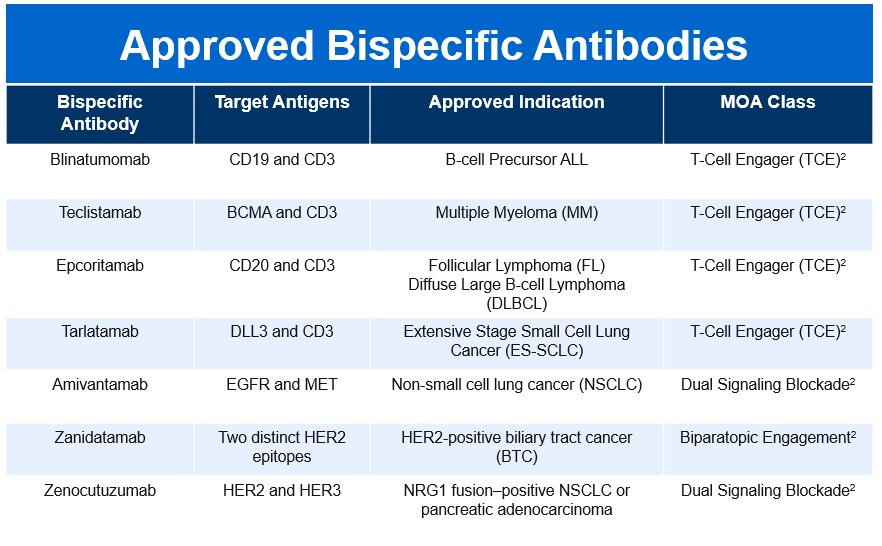
Biomarker-driven therapy has transformed the treatment landscape for metastatic GEA. Approximately 14 to 22% of GEAs are HER2-positive [3-5], and HER2-targeted therapy has become central to treatment. First line therapy for advanced GEA currently consists of FOLFOX plus trastuzumab, with the addition of pembrolizumab for PD-L1 positive (combined positive score ≥ 1) disease, based on the Keynote-811 trial [6]. This trial showed a median overall survival (OS) of 20 months for pembrolizumab added to trastuzumab and chemotherapy, versus 16.8 months for trastuzumab and chemotherapy alone. Median progression-free survival (PFS) was 10.0 versus 8.1 months, respectively [6]. HER2-directed therapy remains relevant in the second line setting, with the antibody-drug conjugate trastuzumab deruxtecan (T-Dxd) available. While these treatments reflect impactful progress, there remains a critical need for advanced HER2-directed therapies that deliver deeper and more durable responses. Zanidatamab is a dual HER2-targeted bispecific immunoglobulin G1-like antibody that binds to two separate extracellular domains of HER2 (domains 2 and 4) [7]. Zanidatamab has already received accelerated approval for previously treated unresectable or metastatic HER2-positive biliary tract cancer, and is under investigation for the treatment GEA [7].
The ongoing Herizon-GEA-01 study is a phase 3 randomized trial evaluating first-line zanidatamab in combination with chemotherapy, plus or minus anti-PD-1 immunotherapy [8]. Participants were 18 years or older with histologically confirmed unresectable, locally advanced, recurrent or metastatic HER2-positive GEA [8]. HER2 positivity consisted of a HER2 immunohistochemistry (IHC) score of 3+, or IHC2+ with positive in situ hybridization. Other inclusion criteria were an ECOG performance status of 0 or 1, assessable disease defined by RECIST 1.1 criteria, adequate organ function, and left ventricular ejection fraction at least 50%. Select exclusion criteria were untreated or symptomatic CNS metastases, prior treatment for locally advanced/metastatic GEA, prior HER2-targeted treatment, and prior immunotherapy including anti-PD-1 and anti-PD-L1 agents.
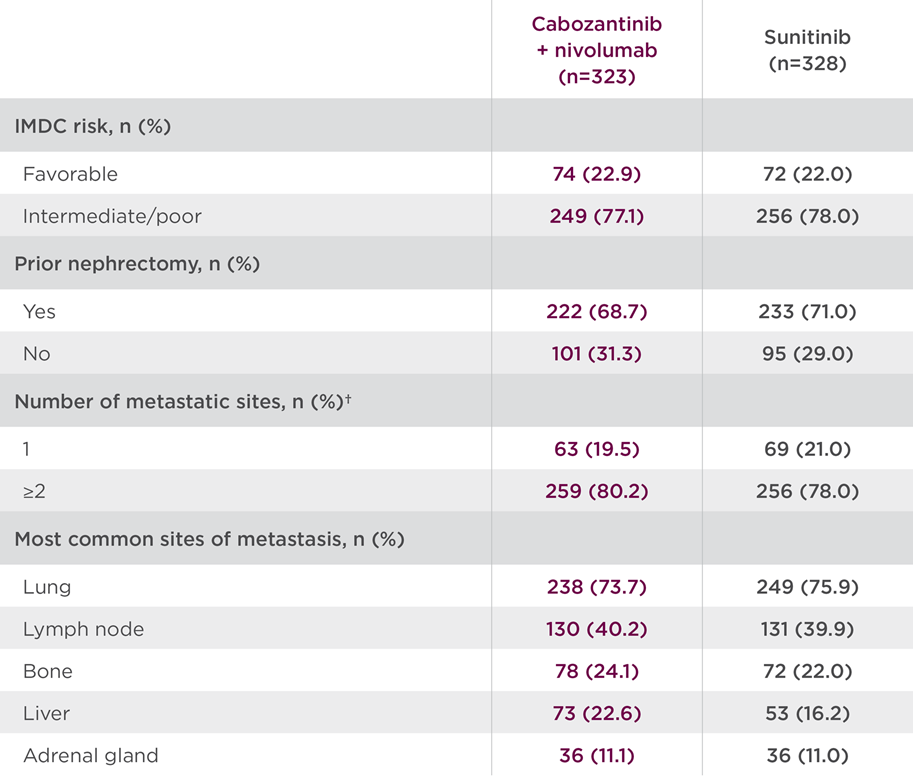
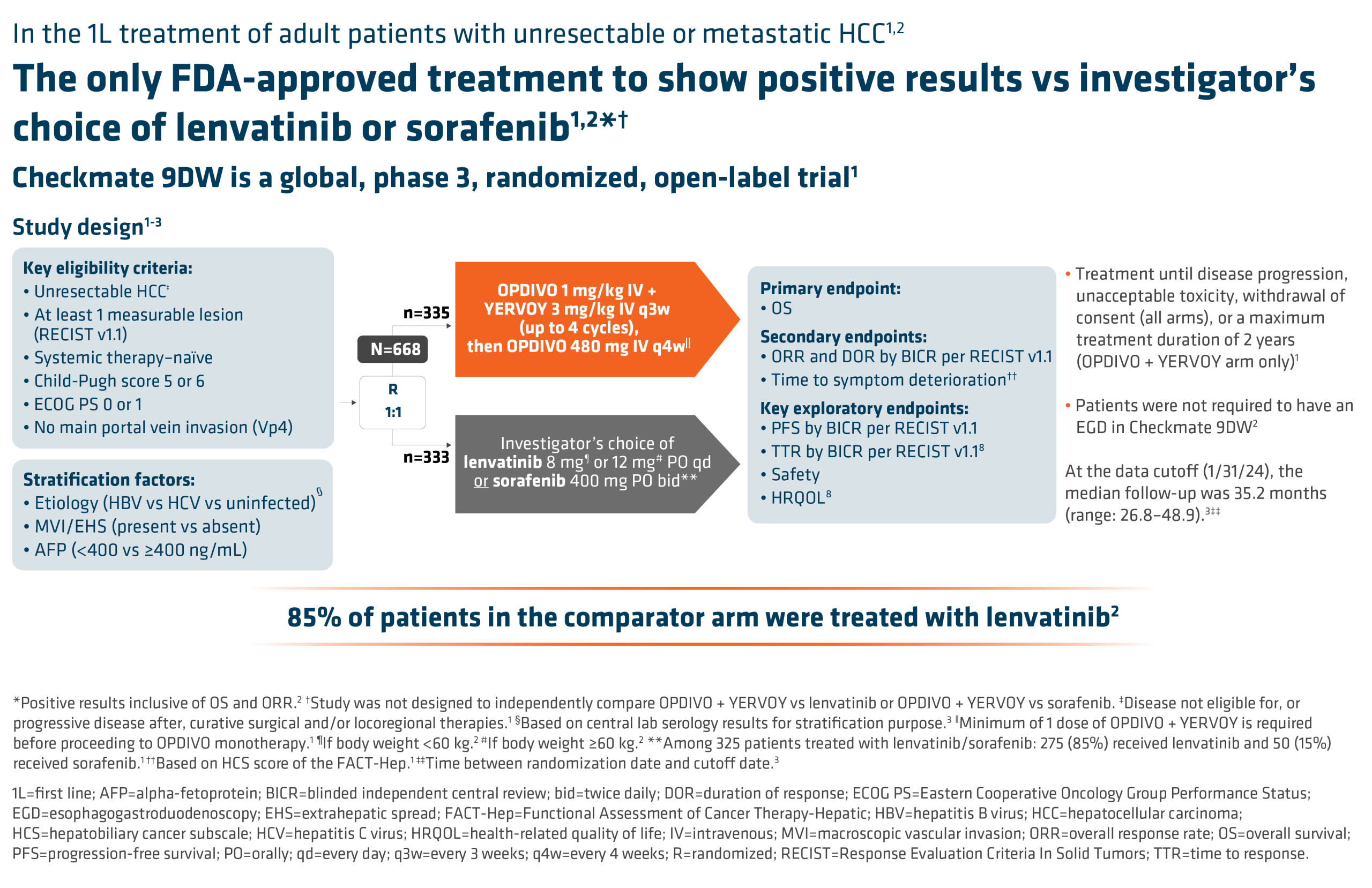
In Herizon-GEA-01, 914 patients were stratified based on geographic region, HER2 status, and ECOG performance status. Participants were randomly assigned 1:1:1 to one of three treatment arms [8,9]. Arm A patients received trastuzumab plus physician’s choice of chemotherapy (CAPOX or FP). Arm B received IV zanidatamab 1800mg (weight <70kg)/2400mg (weight ≥70kg) Q3W plus physician’s choice of chemotherapy. Arm C received IV zanidatamab plus IV tislelizumab 200mg Q3W plus physician’s choice of chemotherapy.
Patients continued treatment until death, disease progression, or unacceptable toxicity. Chemotherapy could be discontinued after 6 cycles. The median age in the treatment arms ranged from 62.5 to 64. Most patients were male (77.3 to 80.8%), ECOG performance status of 1 (55.9 to 61%), metastatic disease (94 to 97.1%), HER2 IHC 3+ (82.6 to 83.1%) and with a PD-L1 tumor area positivity (TAP) score of 1% or greater (58.6 to 61.9%) [10]. Dual primary endpoints were PFS by blinded independent central review and OS. Secondary endpoints included objective response rate, duration of response, safety, health-related quality of life, and pharmacokinetics.
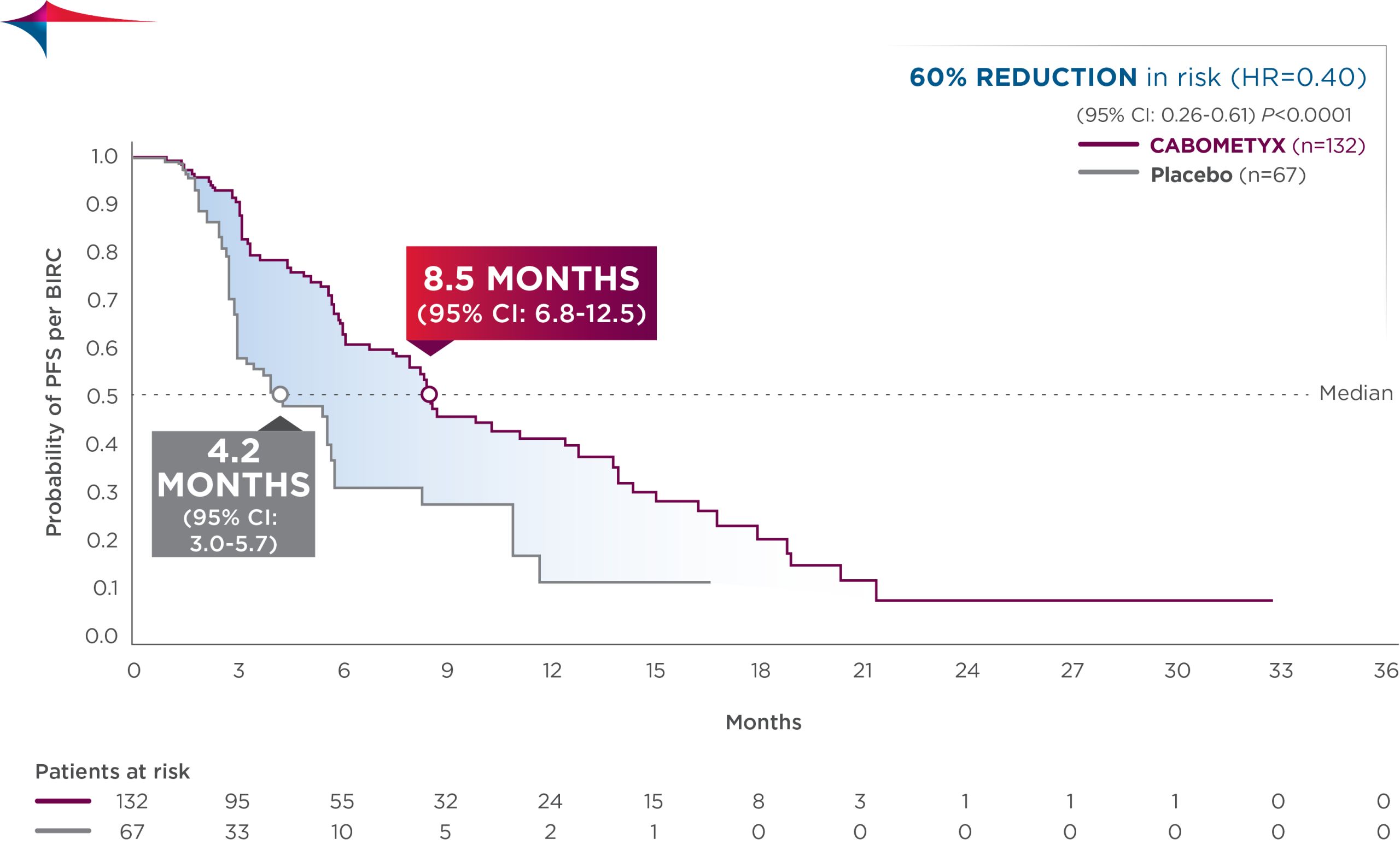
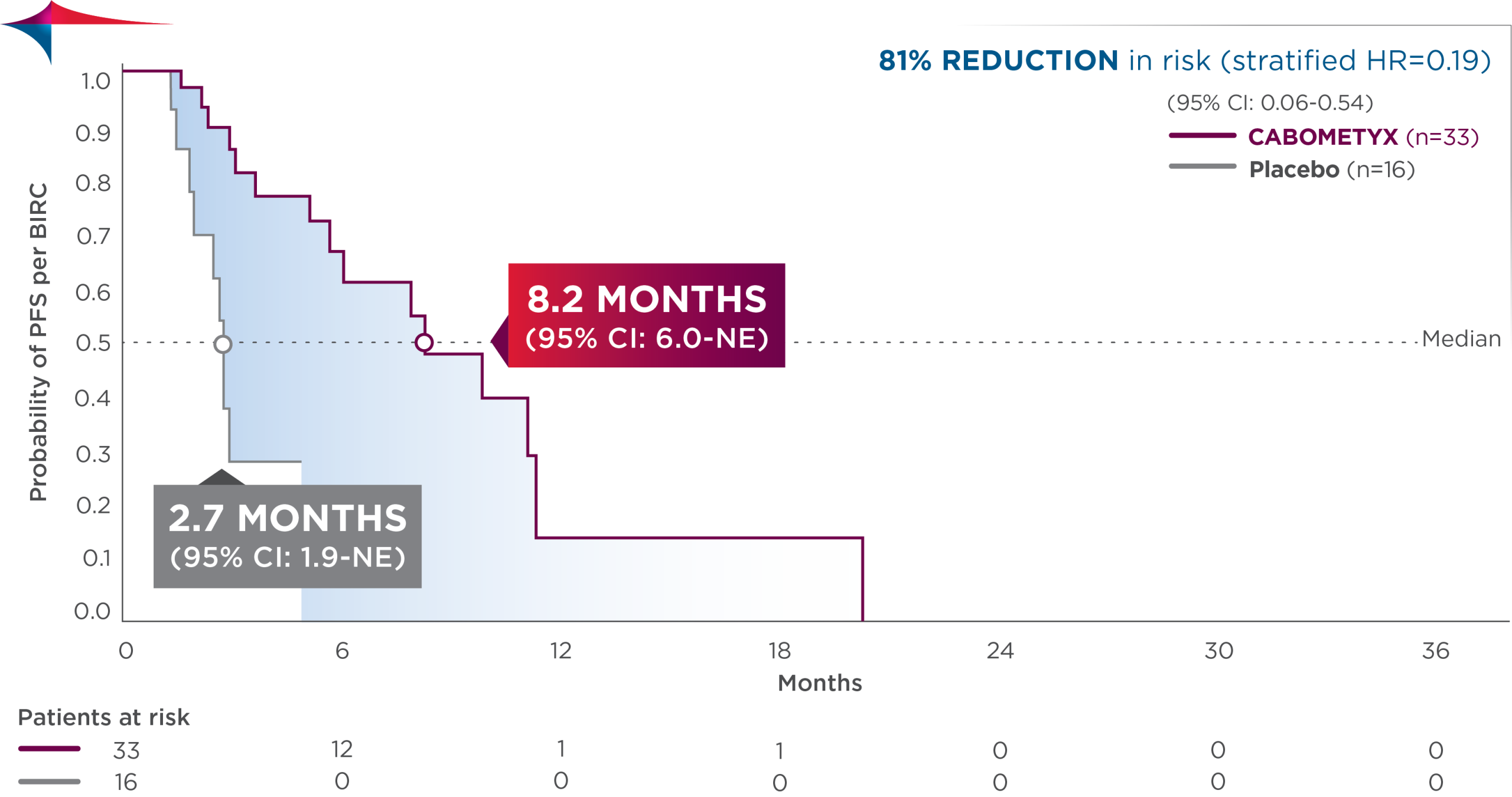
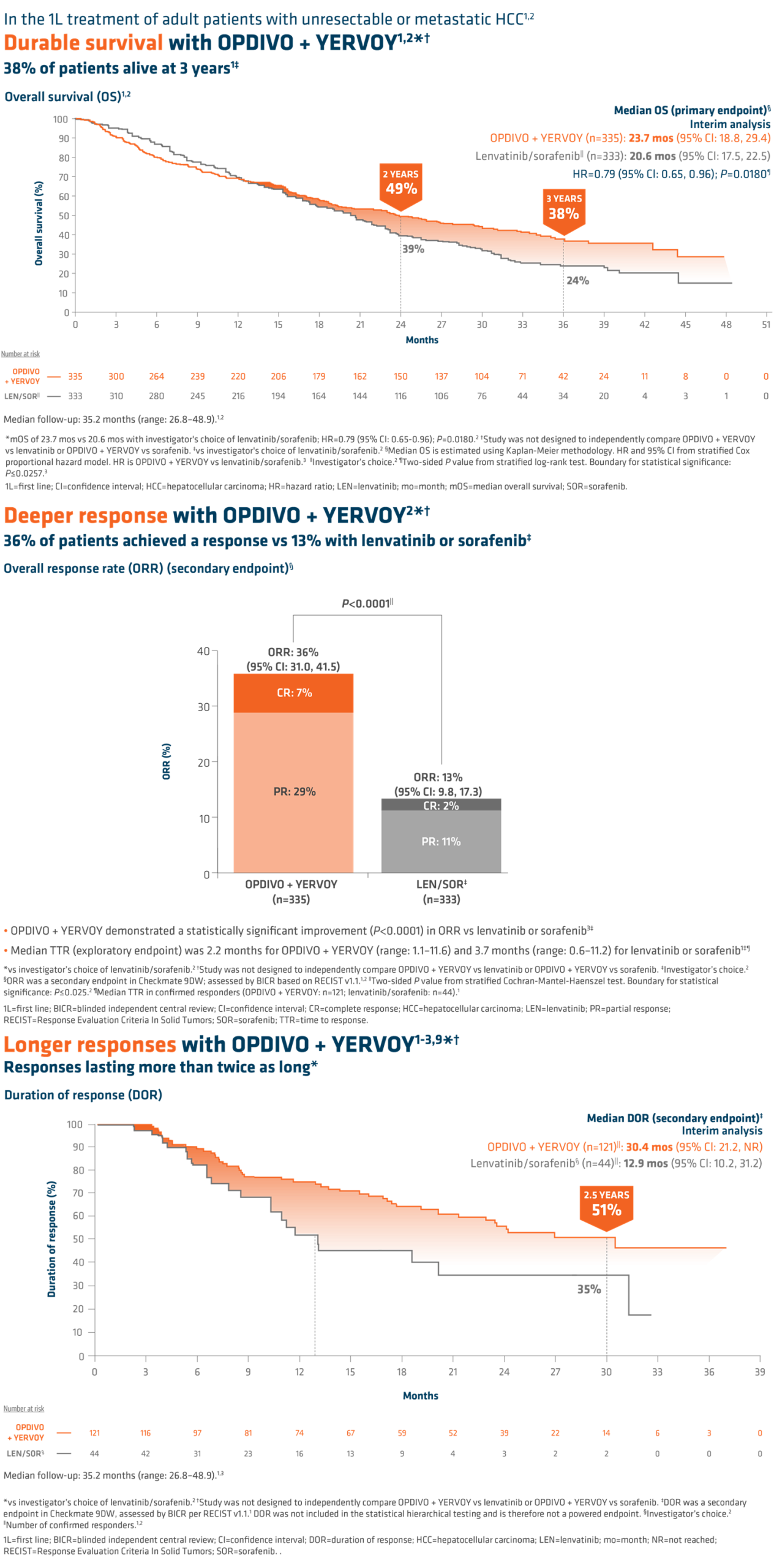
Interim analysis of HERIZON-GEA-01 demonstrates deeper and more durable responses in the zanidatamab arms than those seen in the trastuzumab plus chemotherapy control arm [10,11]. The median PFS was 12.4 months in both zanidatamab-treated arms compared to 8.1 months in the control arm. Median overall survival was 24.4 months in the zanidatamab plus chemotherapy arm, and 26.4 in the zanidatamab plus tislelizumab plus chemo arm, compared to 19.2 months in the control arm. Objective response rates were 65.7% in the control arm, 69.6% in the zanidatamab plus chemotherapy arm, and 70.7% in the zanidatamab plus tislelizumab plus chemotherapy arm. Median duration of response was 8.3 months in the control arm, 14.3 months in the zanidatamab plus chemotherapy arm, and 20.7 months in the zanidatamab plus tislelizumab plus chemotherapy arm.
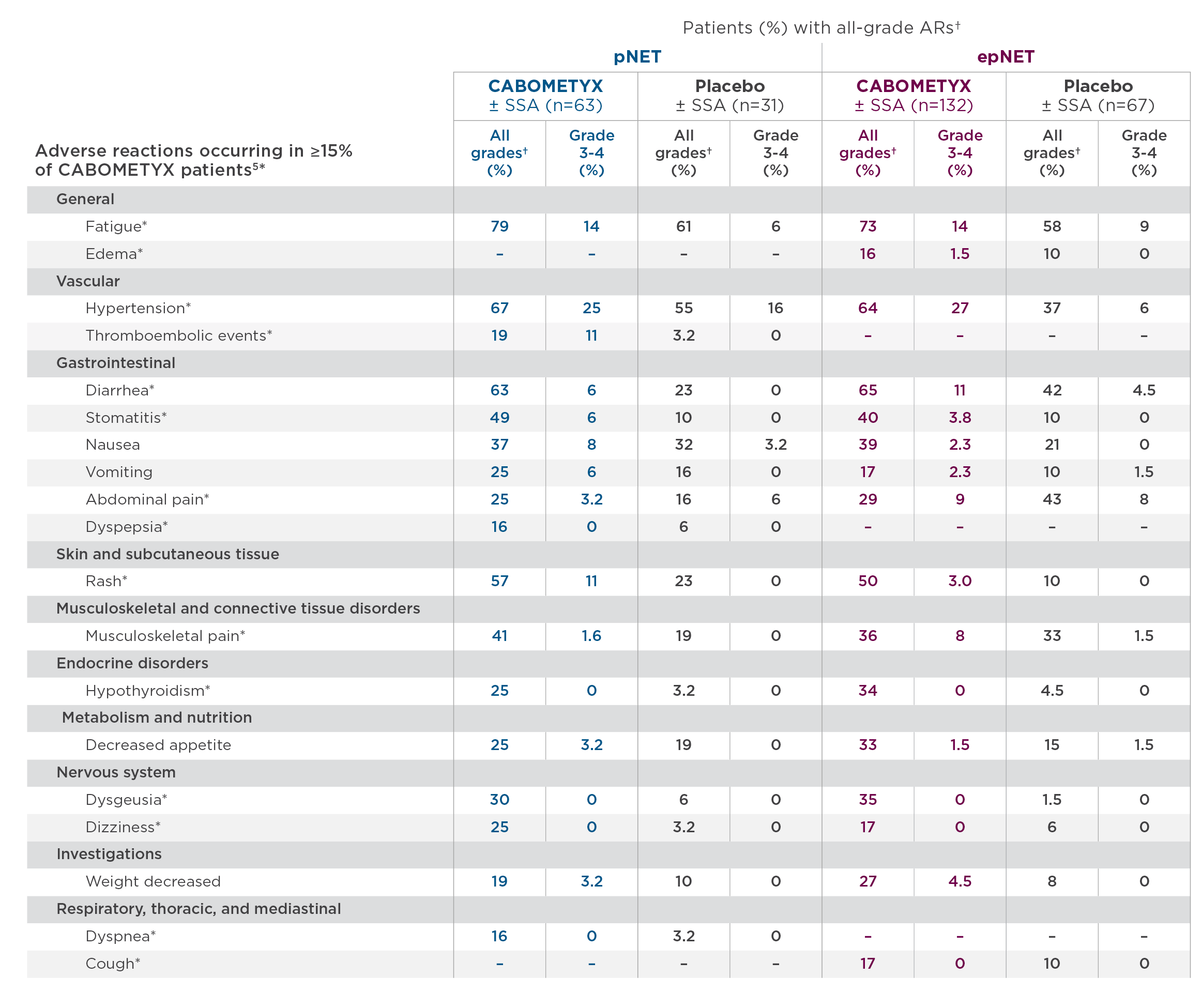
Safety data from HERIZON-GEA-01 interim analysis reveals expected results and a manageable zanidatamab toxicity profile. Diarrhea was the most common adverse event across all three arms, occurring in 82% and 76% of patients in the zanidatamab-treated arms [10]. Other common adverse events across all groups included nausea, vomiting and decreased appetite [11]. Of note, diarrhea prophylaxis was mandatory for patients treated with zanidatamab. Therefore, appropriate antidiarrheal prophylaxis in the clinic will be very important. This trial is ongoing and we anticipate further survival data in the future.
This pivotal data from HERIZON-GEA-01 is anticipated to have a meaningful impact on this patient population. This is particularly in light of the deeper and more long-lasting responses. Over time, we will see if this translates into statistically significant overall survival and progression-free survival benefits.
References:
- Cancer Stat Facts: Stomach Cancer. National Cancer Institute. Surveillance, Epidemiology, and End Results Program. https://seer.cancer.gov/statfacts/html/stomach.html. Accessed May 13, 2026.
- Cancer Stat Facts: Esophageal Cancer. National Cancer Institute. Surveillance, Epidemiology, and End Results Program. https://seer.cancer.gov/statfacts/html/esoph.html. Accessed May 13, 2026.
- Van Cutsem E, Bang YJ, Feng-Yi F, et al. HER2 screening data from ToGA: targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer. 2015;18(3):476-484. doi:10.1007/s10120-014-0402-y.
- Janjigian YY, Werner D, Pauligk C, et al. Prognosis of metastatic gastric and gastroesophageal junction cancer by HER2 status: a European and USA International collaborative analysis. Ann Oncol. 2012;23(10):2656-2662. doi:10.1093/annonc/mds104.
- Kim WH, Gomez-Izquierdo L, Vilardell F, et al. HER2 Status in Gastric and Gastroesophageal Junction Cancer: Results of the Large, Multinational HER-EAGLE Study. Appl Immunohistochem Mol Morphol. 2018;26(4):239-245. doi:10.1097/PAI.0000000000000423.
- Janjigian YY, Kawazoe A, Bai Y, et al. Pembrolizumab in HER2-Positive Gastric Cancer. N Engl J Med. 2024;391(14):1360-1362. doi:10.1056/NEJMc2408121.
- Ziihera ® (zanidatamab-hrii) | Official Website for US Healthcare Professionals. Jazz Pharmaceuticals. https://www.ziiherahcp.com/. Accessed May 13, 2026.
- A Study of Zanidatamab in Combination With Chemotherapy With or Without Tislelizumab in Subjects With HER2-positive Unresectable Locally Advanced or Metastatic Gastroesophageal Adenocarcinoma (GEA). gov. https://clinicaltrials.gov/study/NCT05152147. Accessed May 13, 2026.
- Tabernero J, Shen L, Elimova E, et al. HERIZON-GEA-01: Zanidatamab + chemo ± tislelizumab for 1L treatment of HER2-positive gastroesophageal adenocarcinoma. Future Oncol. 2022;18(29):3255-3266. doi:10.2217/fon-2022-0595.
- Elimova E, Rha SY, Shitara K, et al. Zanidatamab + chemotherapy (CT) ± tislelizumab for first-line (1L) HER2-positive (HER2+) locally advanced, unresectable, or metastatic gastroesophageal adenocarcinoma (mGEA): Primary analysis from HERIZON-GEA-01. J Clin Oncol. 2026;44(2 suppl):LBA285. doi:10.1200/JCO.2026.44.2_suppl.LBA285.
- Zanidatamab With or Without Tislelizumab Yields Clinically Meaningful Survival Benefit in HER2-Positive Advanced Gastroesophageal Adenocarcinoma. ASCO Daily News. https://dailynews.ascopubs.org/do/zanidatamab-without-tislelizumab-yields-clinically-meaningful-survival-benefit-her2. Accessed May 13, 2026.