
Insights from the Phase 3 CABINET trial of CABOMETYX® (cabozantinib) including lung and thymic NET
Written by Munveer Bhangoo, MD
Sponsored by Exelixis, Inc.
Neuroendocrine tumors (NETs) are often characterized as a diverse category of diseases that often have widely differing clinical behavior and outcomes. This behavior is at least in part a reflection of the fact that neuroendocrine cells are scattered through the body. Tumors evolving from neuroendocrine cells vary considerably in terms of location, biological aggressiveness, hormone status, and somatostatin receptor status.1 Taken together, patients with this disease exhibit highly variable clinical outcomes.2 Furthermore, the varied clinical and pathologic features of NETs impact the landscape of clinical trials in this space, with a longstanding need for trials to include a population representative of the disease.3,4
Metastatic neuroendocrine tumors have historically represented an unmet need, particularly for patients experiencing disease progression. In March 2025, the FDA approval of cabozantinib (CABOMETYX®) for the treatment of adult and pediatric patients 12 years of age and older with previously treated, unresectable, locally advanced or metastatic, well-differentiated pancreatic neuroendocrine tumors (pNET) or extrapancreatic neuroendocrine tumors (epNET) marked a meaningful advancement in addressing this gap.3,5 As the only FDA-approved option for patients with previously treated NET, regardless of primary site, tumor grade, SSTR expression, or functional disease, CABOMETYX offers an approved treatment option for patients whose disease progresses on an initial therapy.5-11
Let’s review the pivotal trial data from CABINET, a Phase 3 trial enrolling a heterogeneous population with both pancreatic and extrapancreatic NET, which spanned gastrointestinal, lung, thymus, and unknown and other sites of origin.3
CABINET trial design
CABINET was a randomized (2:1), double-blind, placebo-controlled, Phase 3, National Cancer Institute-sponsored trial of CABOMETYX vs placebo in advanced NET patients previously treated with ≥1 FDA-approved systemic therapy, not including an SSA. CABINET enrolled 2 independent cohorts that evaluated patients with pNET (N=99) or epNET (N=199). The starting dose for CABOMETYX was 60 mg, administered orally once daily. The primary endpoint was PFS; ORR and OS were secondary endpoints.3,5
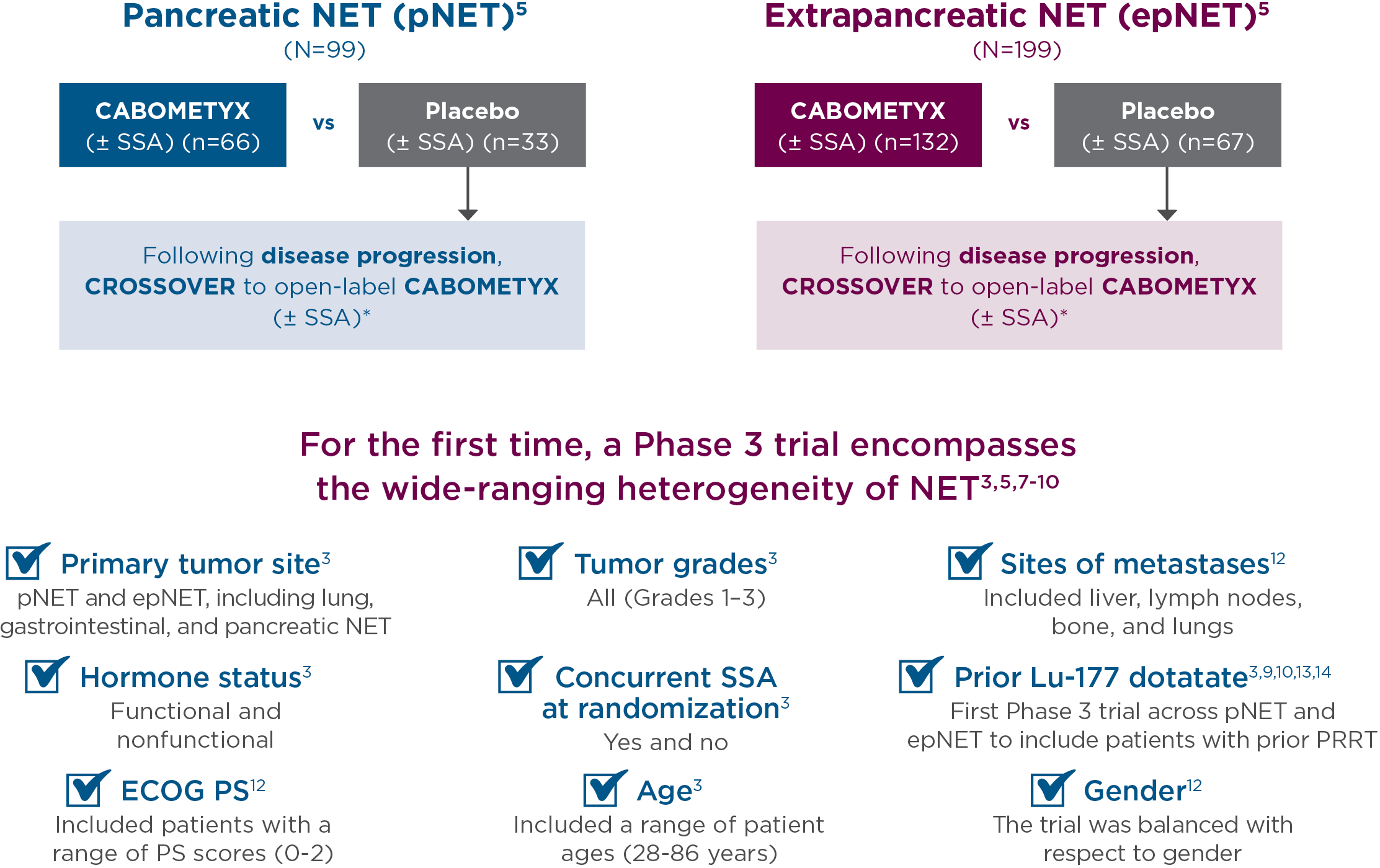
*Unblinding and crossover to open-label CABOMETYX allowed after confirmation of progressive disease by real-time central radiology review.5
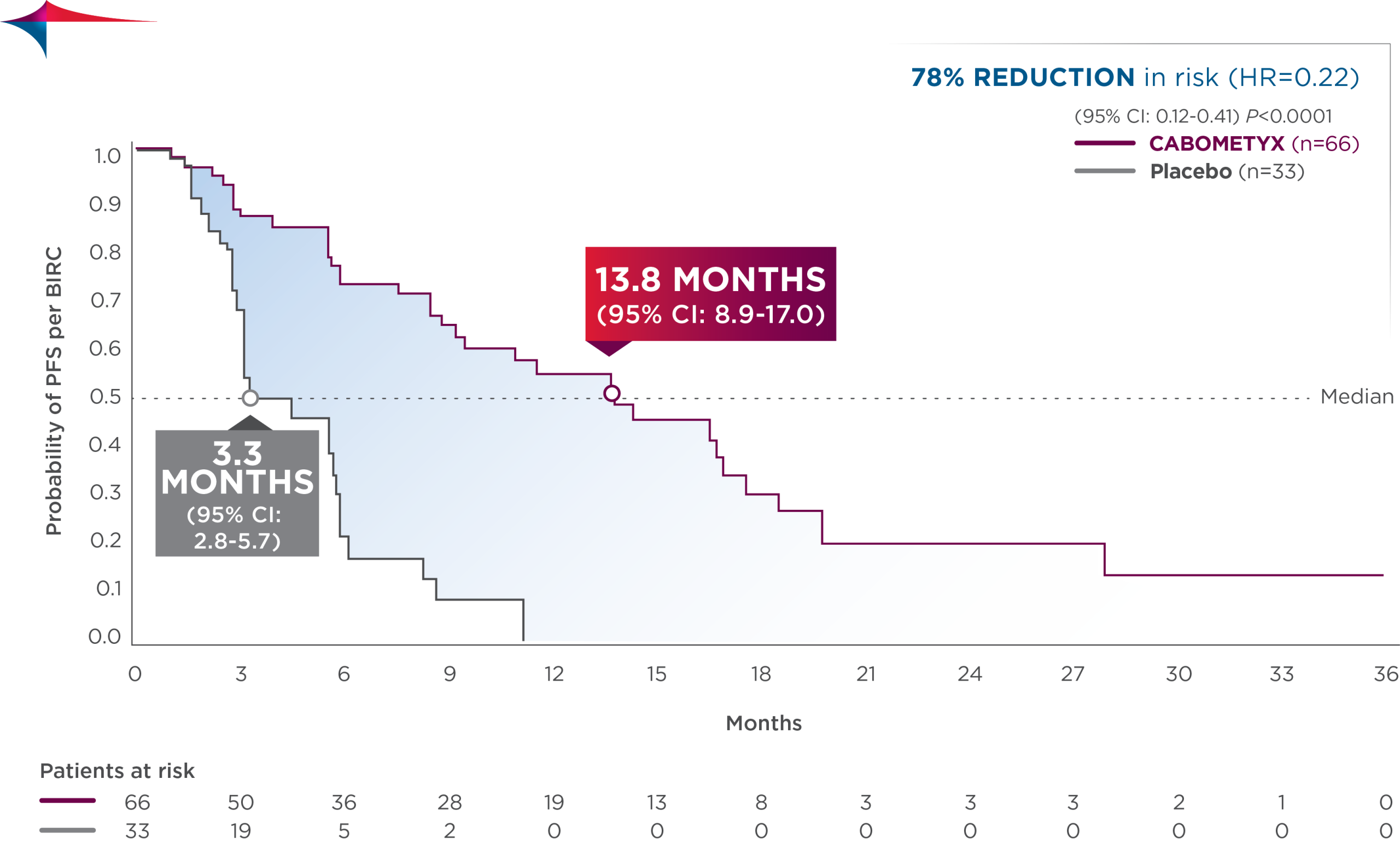
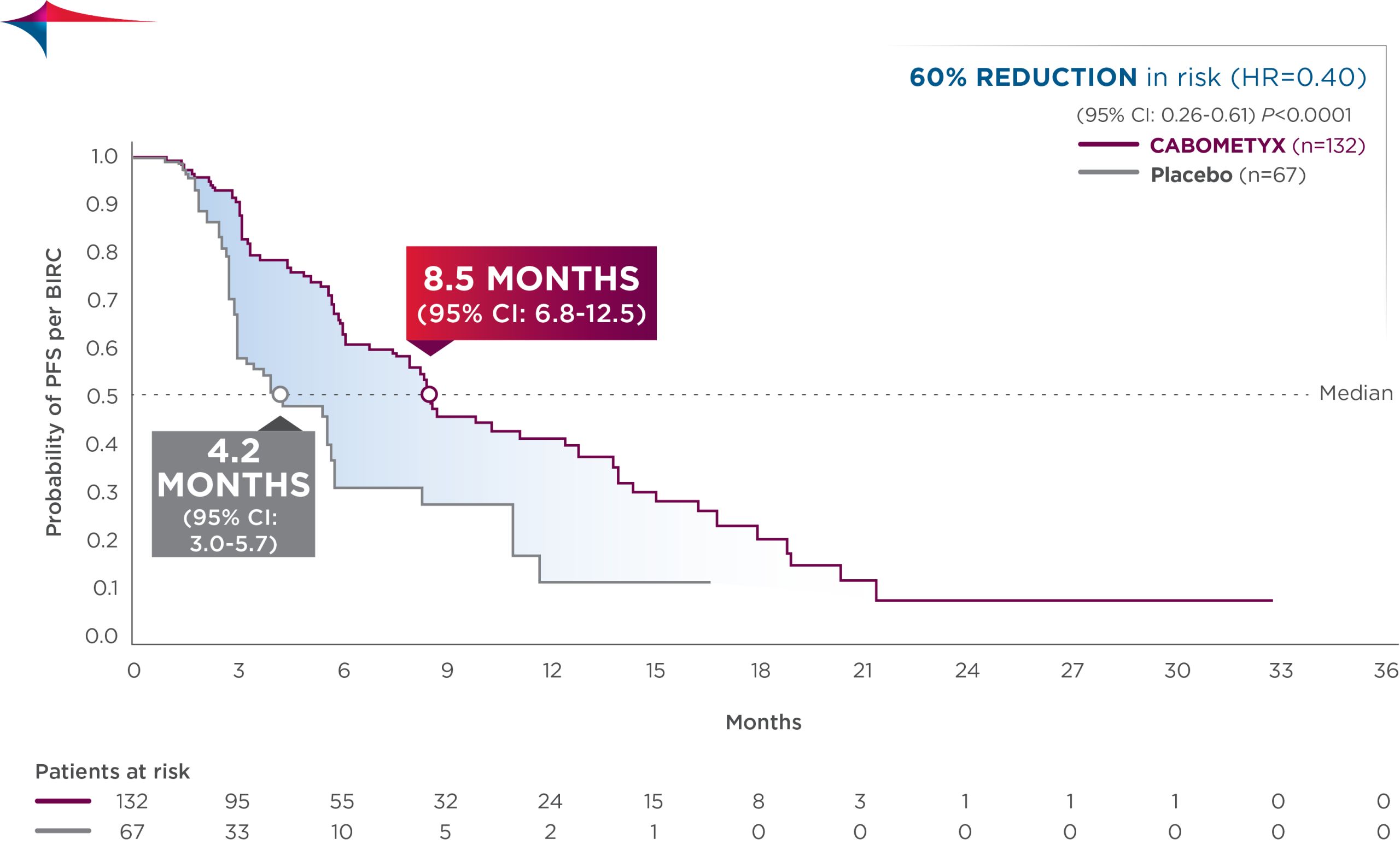
Primary efficacy results in CABINET
CABOMETYX quadrupled median PFS in pNET and doubled median PFS in epNET5
- pNET: median PFS was 13.8 months (95% CI: 8.9-17.0; n=66) vs 3.3 months with placebo (95% CI: 2.8-5.7; n=33); HR=0.22 (95% CI: 0.12-0.41); P<0.0001
- epNET: median PFS was 8.5 months (95% CI: 6.8-12.5; n=132) vs 4.2 months with placebo (95% CI: 3.0-5.7; n=67); HR=0.40 (95% CI: 0.26-0.61); P<0.0001
Exploratory subgroup analysis: PFS results in patients with lung/thymus site origin15
- In the lung /thymus subgroup, median PFS was 8.2 months (95% CI: 6.0-NE; n=33) vs 2.7 months (95% CI: 1.9-NE; n=16) with placebo; HR=0.19 (95% CI: 0.06-0.54)
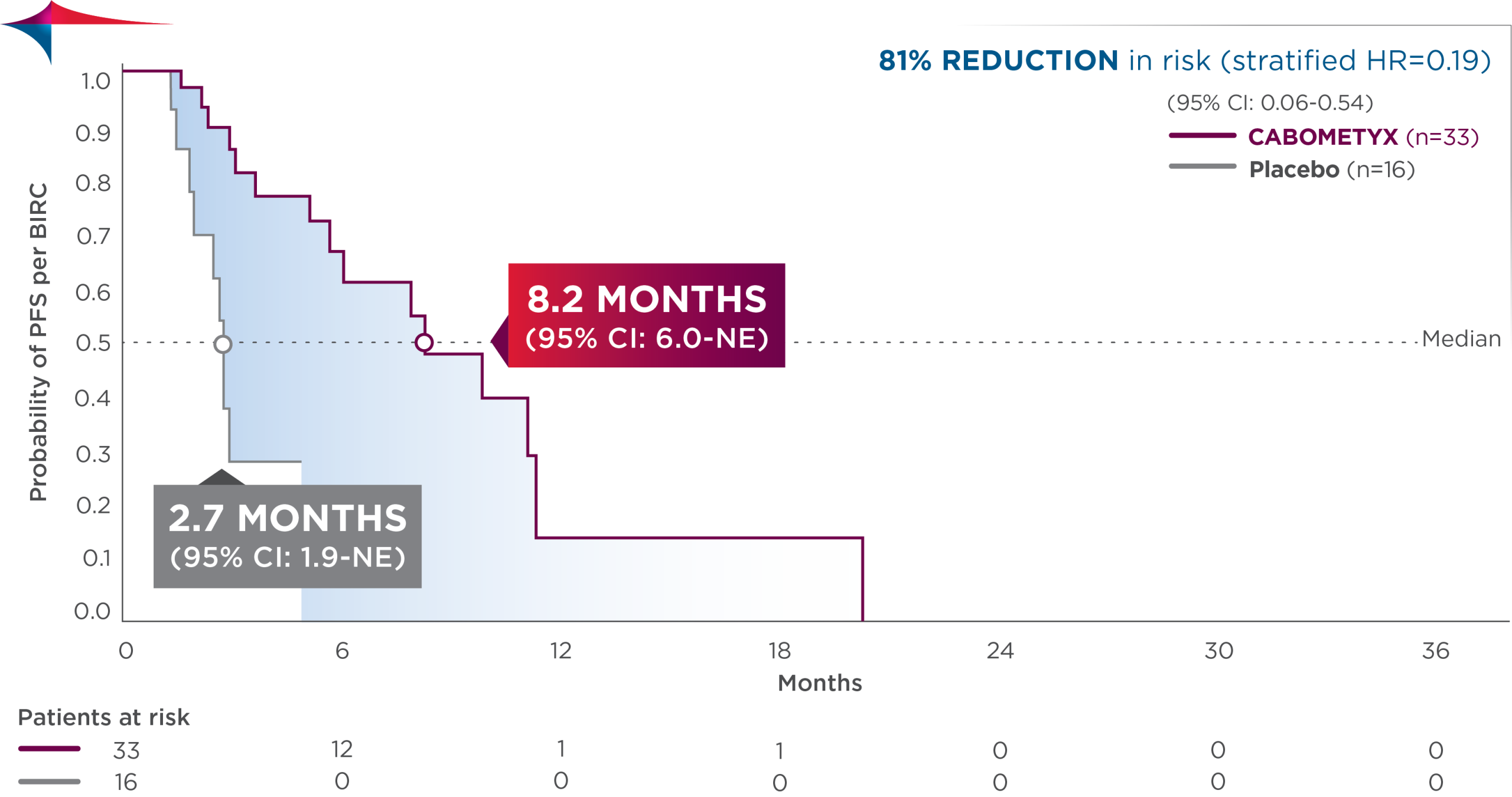
Observed outcomes should be interpreted with caution because of the relatively small subgroup size. Subgroups were not powered to show differences between treatment arms, and results should be considered hypothesis generating.
CABINET safety results
The safety profile observed in CABINET was consistent with the known CABOMETYX safety profile, and no new safety signals were identified in the trial.3
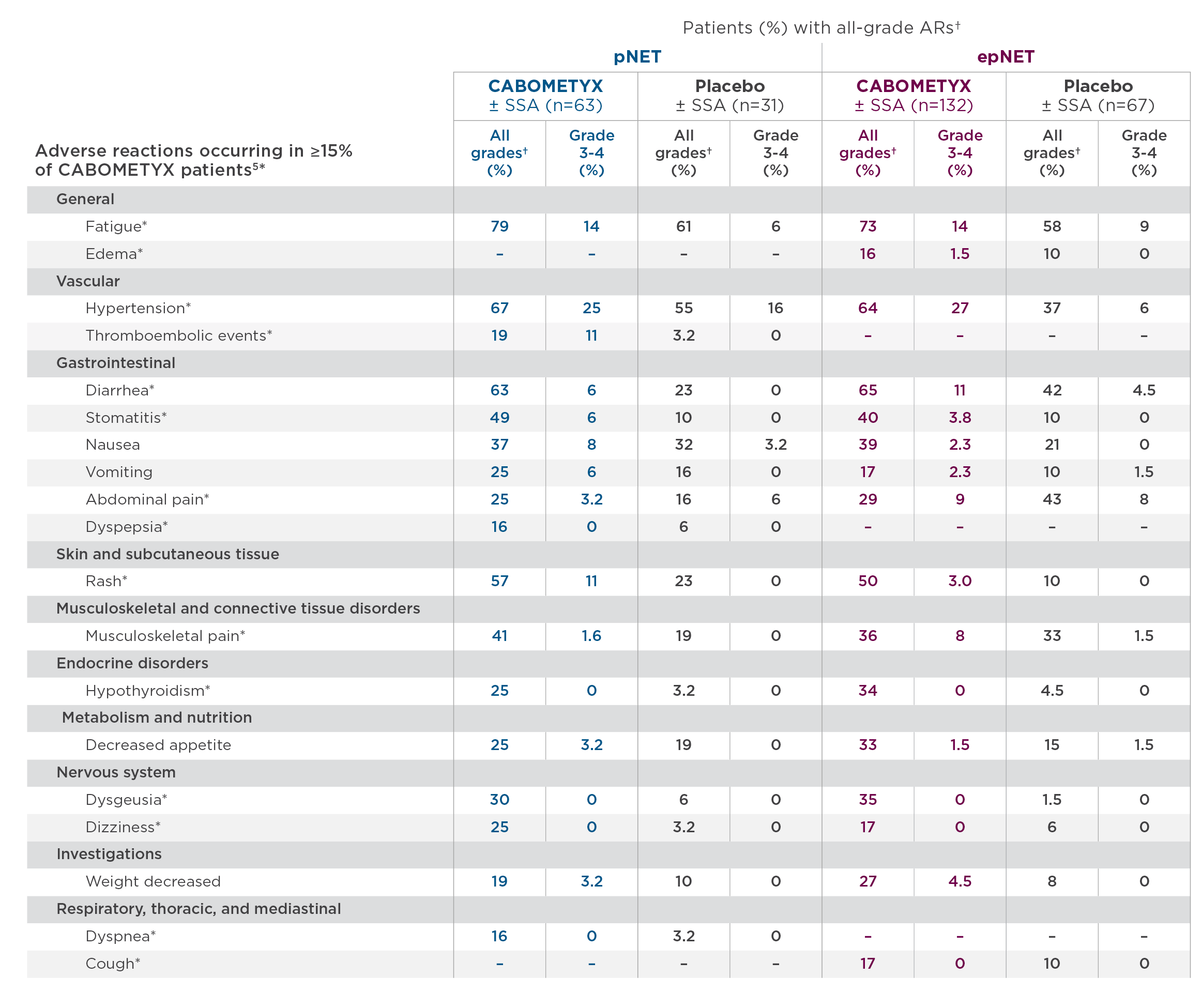
- Commonly occurring treatment-related Grade 3 and 4 adverse events in the lung/thymus subgroup included fatigue, hypertension, diarrhea, and PPE15
*These ARs are grouped terms.5 For details, please see full Prescribing Information.
†NCI CTCAE Version 5.0.
CABOMETYX dosing for NET
The recommended starting dose of CABOMETYX for adult and pediatric patients 12 years of age and older and ≥40 kg is 60 mg once daily until disease progression or unacceptable toxicity (with dose reductions to 40 mg and 20 mg once daily). The recommended starting dose for pediatric patients 12 years of age and older and <40 kg is 40 mg (with dose reductions to 20 mg daily and 20 mg every other day). If previously receiving lowest dose, resume at same dose. If lowest dose not tolerated, discontinue CABOMETYX. The median average daily dose of CABOMETYX treatment was 41 mg in the pNET cohort and 43 mg in the epNET cohort.5
It is important to note that the overall efficacy results of the CABINET trial were achieved in the context of dose modifications.16
- pNET: AR-related dose reductions occurred in 49% of patients receiving CABOMETYX vs 16% with placebo. Discontinuation due to ARs occurred in 19% of patients receiving CABOMETYX vs 10% with placebo
- epNET: AR-related dose reductions occurred in 38% of patients receiving CABOMETYX vs 6% with placebo. Discontinuation due to ARs occurred in 28% of patients receiving CABOMETYX vs 19% receiving placebo
In summary, based on the CABINET data:
Cabozantinib became the first systemic therapy indicated for previously treated NETs regardless of origin based on the landmark Phase 3 CABINET trial.3,5,7-11 Furthermore, the broad eligibility criteria for CABINET allowed for the inclusion of patients irrespective of functional status or expression of somatostatin receptors.3
Taken together, the approval of cabozantinib represents an important advancement in the management of previously treated patients with advanced NETs across diverse sites of origin, including those with lung and thymic origin.3,5,17
Dr Bhangoo received a fee for participating in the development of this article, and his comments reflect his opinions and are not intended to constitute medical advice for individual patients
AR=adverse reaction; CI=confidence interval; DCR=disease control rate; ECOG PS=Eastern Cooperative Oncology Group performance status; epNET=extrapancreatic neuroendocrine tumor; HR=hazard ratio; NET=neuroendocrine tumor; ORR=overall response rate; OS=overall survival; NE=not estimable; PET=positron emission tomography; PFS=progression-free survival; pNET=pancreatic neuroendocrine tumor; PPE=palmar-plantar erythrodysesthesia; PRRT=peptide receptor radionuclide therapy.
INDICATIONS
CABOMETYX® (cabozantinib) is indicated for the treatment of adult and pediatric patients 12 years of age and older with previously treated, unresectable, locally advanced or metastatic, well-differentiated pancreatic neuroendocrine tumors (pNET).
CABOMETYX is indicated for the treatment of adult and pediatric patients 12 years of age and older with previously treated, unresectable, locally advanced or metastatic, well-differentiated extrapancreatic neuroendocrine tumors (epNET).
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Hemorrhage: CABOMETYX can cause severe and fatal hemorrhages. The incidence of Grade 3-5 hemorrhagic events was 5% in CABOMETYX patients in RCC, HCC, and DTC studies. Discontinue CABOMETYX for Grade 3-4 hemorrhage and before surgery. Do not administer to patients who have a recent history of hemorrhage, including hemoptysis, hematemesis, or melena.
Perforations and Fistulas: Fistulas, including fatal cases, and gastrointestinal (GI) perforations, including fatal cases, each occurred in 1% of CABOMETYX patients. Monitor for signs and symptoms, and discontinue CABOMETYX in patients with Grade 4 fistulas or GI perforation.
Thromboembolic Events: CABOMETYX can cause arterial or venous thromboembolic events. Venous thromboembolism occurred in 7% (including 4% pulmonary embolism) and arterial thromboembolism in 2% of CABOMETYX patients. Fatal thrombotic events have occurred. Discontinue CABOMETYX in patients who develop an acute myocardial infarction or serious arterial or venous thromboembolic events.
Hypertension and Hypertensive Crisis: CABOMETYX can cause hypertension, including hypertensive crisis. Hypertension was reported in 37% (16% Grade 3 and <1% Grade 4) of CABOMETYX patients. In CABINET (n=195), hypertension occurred in 65% (26% Grade 3) of CABOMETYX patients. Do not initiate CABOMETYX in patients with uncontrolled hypertension. Monitor blood pressure regularly during CABOMETYX treatment. Withhold CABOMETYX for hypertension that is not adequately controlled; when controlled, resume at a reduced dose. Permanently discontinue CABOMETYX for severe hypertension that cannot be controlled with antihypertensive therapy or for hypertensive crisis.
Cardiac Failure: CABOMETYX can cause severe and fatal cardiac failure. Cardiac failure occurred in 0.5% of patients treated with CABOMETYX as a single agent, including fatal cardiac failure in 0.1% of patients. Consider baseline and periodic evaluations of left ventricular ejection fraction. Monitor for signs and symptoms of cardiovascular events. Withhold and resume at a reduced dose upon recovery or permanently discontinue depending on the severity.
Diarrhea: CABOMETYX can cause diarrhea and it occurred in 62% (10% Grade 3) of treated patients. Monitor and manage patients using antidiarrheals as indicated. Withhold CABOMETYX until improvement to ≤ Grade 1; resume at a reduced dose.
Palmar-Plantar Erythrodysesthesia (PPE): CABOMETYX can cause PPE and it occurred in 45% of treated patients (13% Grade 3). Withhold CABOMETYX until PPE resolves or decreases to Grade 1 and resume at a reduced dose for intolerable Grade 2 PPE or Grade 3 PPE.
Proteinuria: Proteinuria was observed in 8% of CABOMETYX patients. Monitor urine protein regularly during CABOMETYX treatment. For Grade 2 or 3 proteinuria, withhold CABOMETYX until improvement to ≤ Grade 1 proteinuria; resume CABOMETYX at a reduced dose. Discontinue CABOMETYX in patients who develop nephrotic syndrome.
Osteonecrosis of the Jaw (ONJ): CABOMETYX can cause ONJ and it occurred in <1% of treated patients. Perform an oral examination prior to CABOMETYX initiation and periodically during treatment. Advise patients regarding good oral hygiene practices. Withhold CABOMETYX for at least 3 weeks prior to scheduled dental surgery or invasive dental procedures. Withhold CABOMETYX for development of ONJ until complete resolution; resume at a reduced dose.
Impaired Wound Healing: CABOMETYX can cause impaired wound healing. Withhold CABOMETYX for at least 3 weeks prior to elective surgery. Do not administer for at least 2 weeks after major surgery and until adequate wound healing. The safety of resumption of CABOMETYX after resolution of wound healing complications has not been established.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS): CABOMETYX can cause RPLS. Perform evaluation for RPLS and diagnose by characteristic finding on MRI any patient presenting with seizures, headache, visual disturbances, confusion, or altered mental function. Discontinue CABOMETYX in patients who develop RPLS.
Thyroid Dysfunction: CABOMETYX can cause thyroid dysfunction, primarily hypothyroidism, and it occurred in 19% of treated patients (0.4% Grade 3). Assess for signs of thyroid dysfunction prior to the initiation of CABOMETYX and monitor for signs and symptoms during treatment.
Hypocalcemia: CABOMETYX can cause hypocalcemia, with the highest incidence in DTC patients. Based on the safety population, hypocalcemia occurred in 13% of CABOMETYX patients (2% Grade 3 and 1% Grade 4). Monitor blood calcium levels and replace calcium as necessary during treatment. Withhold and resume CABOMETYX at a reduced dose upon recovery or permanently discontinue CABOMETYX depending on severity.
Embryo-Fetal Toxicity: CABOMETYX can cause fetal harm. Advise pregnant women of the potential risk to a fetus and advise females of reproductive potential to use effective contraception during treatment with CABOMETYX and for 4 months after the last dose.
ADVERSE REACTIONS The most common (≥20%) adverse reactions are: CABOMETYX as a single agent: diarrhea, fatigue, PPE, decreased appetite, hypertension, nausea, vomiting, weight decreased, and constipation.
DRUG INTERACTIONS
Strong CYP3A4 Inhibitors: If coadministration with strong CYP3A4 inhibitors cannot be avoided, reduce the CABOMETYX dosage. Avoid grapefruit or grapefruit juice.
Strong or Moderate CYP3A4 Inducers: If coadministration with strong or moderate CYP3A4 inducers cannot be avoided, increase the CABOMETYX dosage. Avoid St. John’s wort.
USE IN SPECIFIC POPULATIONS
Lactation: Advise women not to breastfeed during CABOMETYX treatment and for 4 months after the final dose.
Hepatic Impairment: In patients with moderate hepatic impairment, reduce the CABOMETYX dosage. Avoid CABOMETYX in patients with severe hepatic impairment.
Pediatric Use: Physeal widening has been observed in children with open growth plates when treated with CABOMETYX. Physeal and longitudinal growth monitoring is recommended in children (12 years and older) with open growth plates. Consider interrupting or discontinuing CABOMETYX if abnormalities occur. The safety and effectiveness of CABOMETYX in pediatric patients less than 12 years of age have not been established.
Please see accompanying full Prescribing Information by clicking here.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.
References:
1. Sultana Q, Kar J, Verma A, et al. A comprehensive review on neuroendocrine neoplasms: presentation, pathophysiology and management. J Clin Med. 2023;12(15):5138
2. Dasari A, Wallace K, Halperin DM, et al. Epidemiology of neuroendocrine neoplasms in the US. JAMA Netw Open. 2025;8(6):e2515798. doi:10.1001/jamanetworkopen.2025.15798.
3. Chan JA, Geyer S, Zemla T, et al. Phase 3 trial of cabozantinib to treat advanced neuroendocrine tumors. N Engl J Med. 2024; Published online September 16, 2024. doi:10.1056/NEJMoa2403991.
4. Yang K, Li J, Cheng Y, Bai C. Evolving landscape of clinical trials in gastroenteropancreatic neuroendocrine neoplasms in the past two decades. Endocr Connect. 2023;12(4): e220441. doi:10.1530/EC-22-0441.
5. CABOMETYX® (cabozantinib) Prescribing Information. Exelixis, Inc.
6. Bidani K, Marinovic AG, Moond V, Harne P, Broder A, Thosani N. Treatment of pancreatic neuroendocrine tumors: beyond traditional surgery and targeted therapy. J Clin Med. 2025;14(10):3389. doi:10.3390/jcm14103389.
7. LUTATHERA® (lutetium Lu-177 dotatate) Prescribing Information. Novartis Pharmaceuticals Corporation.
8. AFINITOR® (everolimus) Prescribing Information. Novartis Pharmaceuticals Corporation.
9. SUTENT® (sunitinib malate) Prescribing Information. Pfizer, Inc.
10. SOMATULINE® DEPOT (lanreotide) Prescribing Information. Ipsen Pharma Biotech.
11. SANDOSTATIN® LAR DEPOT (octreotide acetate) Prescribing Information. Novartis Pharmaceuticals Corporation.
12. Chan JA, Geyer S, Zemla T, et al. Phase 3 trial of cabozantinib in previously treated advanced neuroendocrine tumors [supplementary appendix]. N Engl J Med. 2024; Published online September 16, 2024. doi:10.1056/ NEJMoa2403991.
13. US Food and Drug Administration. FDA approves new treatment for certain digestive tract cancers. January 26, 2018. Accessed September 5, 2024. https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-certain-digestive-tract-cancers. 14. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
15. Wolin EM, Zemla T, Strosberg JR, et al. Efficacy and safety of cabozantinib for advanced lung and thymus neuroendocrine tumors after progression on prior therapy: subgroup analysis of phase 3 CABINET trial (Alliance A021602). Poster presented at European Society for Medical Oncology Congress; October 17-21, 2025.
16. Data on file. Exelixis, Inc.
17. Oronsky N, Ma PC, Morgensztern D, Carter CA. Nothing but NET: a review of neuroendocrine tumors and carcinomas. Neoplasia. 2017;19(12):991-1002.
©2026 Exelixis, Inc. CA‐3900 04/26