
The FDA on November 30, 2015 approved EMPLICITI® in combination with Lenalidomide and Dexamethasone for the treatment of patients with Multiple Myeloma who have received one to three prior therapies. EMPLICITI® is a product of Bristol-Myers Squibb Company.
Author: RR
Interim PET Scan may Define Prognosis and Provide Treatment Guidance for Patients with Advanced Hodgkin Lymphoma
SUMMARY: The American Cancer Society estimates that in the United States for 2016, about 8500 new cases of Hodgkin lymphoma will be diagnosed and over 1100 patients will die of the disease. Hodgkin lymphoma is classified into two main groups – Classical Hodgkin lymphomas and Nodular Lymphocyte Predominant type, by the World Health Organization. The Classical Hodgkin lymphomas include Nodular sclerosing, Mixed cellularity, Lymphocyte rich, Lymphocyte depleted, subtypes and accounts for approximately 10% of all malignant lymphomas. Nodular sclerosis Hodgkin lymphoma histology, accounts for approximately 80% of Hodgkin lymphoma cases in older children and adolescents in the United States. Classical Hodgkin Lymphoma is a malignancy of primarily B lymphocytes and is characterized by the presence of large mononucleated Hodgkin and giant multinucleated Reed-Sternberg (RS) cells collectively known as Hodgkin and Reed-Sternberg cells (HRS).
Advanced-stage (stage III to stage IV) Classical Hodgkin lymphoma has a cure rate of approximately 70-80% when treated in the first-line setting with a combination of Doxorubicin, Bleomycin, Vinblastine, and Dacarbazine (ABVD regimen). This regimen which was developed more than 40 years ago is less expensive, easy to administer, is generally well tolerated and is often used in first line setting. Nonetheless, this regimen which contains Bleomycin can cause pulmonary toxicity, the incidence of which is higher in older patients and in those who receive consolidation radiotherapy to the thorax. The second most often used regimen in the first-line setting, e-BEACOPP (escalated doses of Bleomycin, Etoposide, Doxorubicin, Cyclophosphamide, Vincristine, Procarbazine, and Prednisone) has been associated with a higher Progression Free Survival as well as higher 5-year Overall Survival (approximately 90%). This regimen however is associated with short and long term toxicities such as prolonged fatigue, permanent fertility, Myelodysplasia and secondary malignancies.
A retrospective study by Gallamini and co-workers in 2007 had shown that a PET scan after two cycles of ABVD chemotherapy was an independent prognostic factor, with a 2-year Progression Free Survival rate of 95%, for those patients with negative interim PET scan, compared to only 12.8% for those with persistently positive PET scan. Based on these observations, the authors in this prospective trial evaluated the benefit of a “response-adapted” approach, by performing a PET scan following 2 cycles of ABVD treatment and modifying therapy based on the interim PET scan findings.
In this randomized controlled trial, 1203 eligible patients with newly diagnosed advanced Classical Hodgkin lymphoma were registered. The median age was 33 years. Following 2 cycles of chemotherapy with ABVD regimen, 1119 patients had an interim PET-CT scan and patients with negative PET findings (83.7%) were randomly assigned in a 1:1 ratio to continue ABVD regimen (ABVD group) or receive ABVD omitting Bleomycin (AVD group), for cycles 3 through 6. Radiotherapy was not recommended for patients with negative findings on interim PET scans. Patients with positive interim PET scan findings following two cycles of ABVD (16%), received 4-6 cycles of BEACOPP regimen. The primary outcome was the difference in the 3-year Progression Free Survival rate between randomized groups.
With a median follow up of 41 months, the 3-year Progression Free Survival was 85.7% with ABVD and 84.4% with AVD and 3 year Overall Survival was 97.2% and 97.6% in these two respective groups. Pulmonary toxicities were more severe in the ABVD group than in the AVD group and deleting Bleomycin following 2 cycles of ABVD, in patients with negative interim PET scan, did not compromise outcomes. Patients who received BEACOPP regimen based on a positive interim PET scan after the first 2 cycles of ABVD (N=172), had a 3-year Progression Free Survival of 67.5% and Overall Survival rate of 87.8%.
The authors concluded that following 2 cycles of ABVD regimen, omitting Bleomycin from the ABVD regimen, based on a negative interim PET scan (response-adapted therapy), resulted in lower incidence of pulmonary toxicities, compared with continued treatment with ABVD, without compromising efficacy. Adapted Treatment Guided by Interim PET-CT Scan in Advanced Hodgkin’s Lymphoma. Johnson P, Federico M, Kirkwood A, et al. N Engl J Med 2016; 374:2419-2429
ASCO Guidelines on Use of Biomarkers in Early Stage Breast Cancer Part 1
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 246,660 new cases of invasive breast cancer will be diagnosed in 2016 and 40,450 women will die of the disease. Patients with early stage breast cancer often receive adjuvant therapy. Tumor biomarker assays have become an integral part of the treatment decision making process along with clinical and histologic tumor characteristics, further enabling customized care for patients with early-stage invasive breast cancer. A multitude of biomarker assays are presently available for the practicing Health Care Provider. Choosing the appropriate biomarker assay for a given patient can be a daunting task and the ASCO guidelines set forth herein, were developed by an expert panel based on systematic reviews, meta-analyses, randomized controlled trials, prospective-retrospective studies, and prospective comparative observational studies, published from 2006 through 2014. These guidelines are only applicable for patients with newly diagnosed, non-metastatic, primary breast cancer, to prognosticate and predict outcomes but they do not however comment on the choice of specific treatment or regimens based on recurrence score. Treatment decisions should take into consideration disease stage, comorbidities and patient preferences. Even though several tests are now recommended in the guidelines, only one test should be used to guide therapy for an individual patient.
Two important questions were addressed by these guidelines – This edition (Part 1) addresses the first Clinical Question
Clinical Question 1: For women with early-stage invasive breast cancer and with known Estrogen receptor/Progesterone receptor and HER2 status, which other biomarkers have demonstrated clinical utility to guide decisions on the need for adjuvant systemic therapy?
Oncotype DX
If a patient has ER/PR positive, HER2 negative, node negative breast cancer, the Oncotype DX 21-gene recurrence score may be used to guide decisions on adjuvant systemic therapy. It should not be used in patients with ER/PR positive, HER2 negative, node positive disease. It should not be used in patients with HER2 positive or triple negative disease.
PAM50 Risk of Recurrence Score
If a patient has ER/PR positive, HER2 negative, node-negative breast cancer, the PAM50 Risk of Recurrence score may be used in conjunction with other clinicopathologic variables to guide decisions on adjuvant systemic therapy. It should not be used in patients with ER/PR positive, HER2 negative, node-positive disease. It should not be used in patients with HER2 positive breast cancer and those with triple-negative breast cancer to guide decisions on adjuvant systemic therapy.
EndoPredict
If a patient has ER/PR positive, HER2 negative, node-negative breast cancer, EndoPredict 12-gene risk score may be used to guide decisions on adjuvant systemic therapy. It should not be used in patients with ER/PR positive, HER2 negative, node-positive disease. It should not be used in patients with HER2 positive or triple-negative disease.
Breast Cancer Index
If a patient has ER/PR positive, HER2 negative, node-negative breast cancer, the Breast Cancer Index may be used to guide decisions on adjuvant systemic therapy. It should not be used in patients with ER/PR positive, HER2 negative, node-positive disease. It should not be used in patients with HER2 positive or triple negative breast cancer to guide decisions on adjuvant systemic therapy.
Urokinase Plasminogen Activator and Plasminogen Activator Inhibitor Type 1
If a patient has ER/PR positive, HER2 negative, node negative breast cancer, Urokinase Plasminogen Activator and Plasminogen Activator Inhibitor Type 1 may be used to guide decisions on adjuvant systemic therapy. It should not be used in patients with HER2 positive or triple negative breast cancer.
MammaPrint
If a patient has ER/PR positive, HER2 negative (node-positive or node-negative) breast cancer, the MammaPrint 70-gene assay should not be used to guide decisions on adjuvant systemic therapy. It should not be used in patients with HER2 positive disease. It should not be used in patients with triple negative breast cancer.
Mammostrat
If a patient has ER/PR positive, HER2 negative (node-positive or node-negative) breast cancer, the Mammostrat 5-protein assay should not be used to guide decisions on adjuvant systemic therapy. It should not be used in patients with HER2 positive or triple negative breast cancer.
Immunohistochemistry 4
If a patient has ER/PR positive, HER2 negative (node-positive or node-negative) breast cancer, Immunohistochemistry 4 (IHC4) should not be used to guide decisions on adjuvant systemic chemotherapy. It should not be used in patients with HER2 positive or triple negative breast cancer.
Circulating Tumor Cells
The clinician should not use circulating tumor cells to guide decisions on adjuvant systemic therapy.
Tumor-Infiltrating Lymphocytes
If a patient has ER/PR positive, HER2 negative (node-positive or node-negative) breast cancer, Tumor-Infiltrating Lymphocytes should not be used for decision making. It should not be used in patients with HER2 positive or triple negative breast cancer.
Protein Encoded by MKI67 Gene
Protein encoded by the MKI67 gene labeling index by IHC should not be used to guide choice on adjuvant chemotherapy.
Extended Endocrine Therapy
If a patient has ER/PR positive, HER2 negative (node-negative) breast cancer and has had 5 years of endocrine therapy without evidence of recurrence, multiparameter gene expression or protein assays (Oncotype DX, EndoPredict, PAM50, Breast Cancer Index, or IHC4) should not be used to guide decisions on extended endocrine therapy.
The Clinical Question 2 will be addressed in the eNL edition (Part 2) next week.
Harris LN, Ismaila N, McShane LM, et al: Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2016;34:1134-1150.
Late Breaking Abstract – ASCO 2016 Liquid Biopsy Can Rapidly Detect Certain Gene Mutations with High Specificity
Late Breaking Abstract – ASCO 2016: Liquid Biopsy Can Rapidly Detect Certain Gene Mutations with High Specificity
SUMMARY: The FDA approved the first “Liquid Biopsy” test on June 1, 2016 for the detection of exon 19 deletions or exon 21 (L858R) substitution mutations in the Epidermal Growth Factor Receptor (EGFR) gene. On the heels of this approval, Zill and colleagues reported the results of the largest liquid biopsy study ever conducted thus far. It has been well established that treatment with EGFR TKIs results in superior outcomes, for patients with tumors harboring exon 19 deletions and exon 21 mutations. The application of precision medicine with targeted therapy requires detection of molecular abnormalities in a tumor specimen, following progression or recurrence. Archived biopsy specimens may not be helpful, as it is important to identify additional mutations in the tumor at the time of recurrence or progression, in order to plan appropriate therapy. Further, recurrent tumors may be inaccessible for a safe biopsy procedure or the clinical condition of the patient may not permit a repeat biopsy. Additionally, the biopsy itself may be subject to sampling error due to tumor heterogeneity. Genotyping cell free DNA in the plasma, also called liquid biopsy, can potentially overcome the shortcomings of repeat biopsies and tissue genotyping, allowing the detection of many more targetable gene mutations, thus resulting in better evaluation of the tumor genome landscape.
The authors in this study utilized Next Generation Sequencing (NGS) of circulating tumor DNA (ctDNA), isolated from plasma specimens (liquid biopsy specimens) of 15,191 patients of whom 37% had advanced lung cancer, 14% had breast cancer, 10% had colorectal cancer and 39% had other malignancies. Seventy genes were targeted and accuracy of ctDNA sequencing was assessed by comparing with matched tissue tests for 386 patients and frequencies of somatic ctDNA alterations per gene were compared to those previously described in tissue sequencing projects such as data from The Cancer Genome Atlas (TCGA).
It was noted that the ctDNA mutation patterns were highly concordant with tissue analysis as reported by the TCGA. The overall accuracy of ctDNA sequencing in comparison with matched tissue tests was 87% and the accuracy increased to 98% when blood and tumor were collected less than six months apart. Pearson Correlation between sets of data is a measure of how well these sets are related. Between 0.5 and 1.0 is considered high correlation. Pearson correlation for TP53 gene was 0.94, for KRAS was 0.99 and for PIK3CA was 0.99.
The researchers commented on the clinical outcome benefits using liquid biopsy, in four distinct groups:
1) Testing for actionable mutations (ALK fusion, EGFR or BRAF activating mutations in lung; ERBB2 amplification in gastric cancer) in cases with insufficient tissue quantity.
2) Testing for actionable resistance mutations (MET amplification or EGFR T790M in lung cancer), at the time of progression.
3) Genomic evolution upon progression such as ERBB2-amplified metastatic breast cancer in patients with triple negative primary tumor.
4) Tumors with genotypes that need more extensive driver mutation testing such as BRAF V600E in lung.
The authors concluded that there is a high correlation between ctDNA plasma samples and tissue testing with the exception of resistance mutations such as EGFR T790M mutation which evolve while on anti-EGFR inhibitor therapy and consequently may not correlate with the TCGA, probably because patients in the tissue-based population had not yet received the anti-EGFR inhibitor therapy that promotes the mutation. Patients who received treatment based on ctDNA findings also experienced better clinical outcomes. Zill OA, Mortimer S, Banks KC, et al Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. J Clin Oncol. 2016;34(suppl; abstr LBA11501).
YONDELIS® – A Marine-Derived Drug for Soft Tissue Sarcomas
SUMMARY: The FDA approved the first “Liquid Biopsy” test on June 1, 2016 for the detection of exon 19 deletions or exon 21 (L858R) substitution mutations in the Epidermal Growth Factor Receptor (EGFR) gene. On the heels of this approval, Zill and colleagues reported the results of the largest liquid biopsy study ever conducted thus far. It has been well established that treatment with EGFR TKIs results in superior outcomes, for patients with tumors harboring exon 19 deletions and exon 21 mutations. The application of precision medicine with targeted therapy requires detection of molecular abnormalities in a tumor specimen, following progression or recurrence. Archived biopsy specimens may not be helpful, as it is important to identify additional mutations in the tumor at the time of recurrence or progression, in order to plan appropriate therapy. Further, recurrent tumors may be inaccessible for a safe biopsy procedure or the clinical condition of the patient may not permit a repeat biopsy. Additionally, the biopsy itself may be subject to sampling error due to tumor heterogeneity. Genotyping cell free DNA in the plasma, also called liquid biopsy, can potentially overcome the shortcomings of repeat biopsies and tissue genotyping, allowing the detection of many more targetable gene mutations, thus resulting in better evaluation of the tumor genome landscape.
The authors in this study utilized Next Generation Sequencing (NGS) of circulating tumor DNA (ctDNA), isolated from plasma specimens (liquid biopsy specimens) of 15,191 patients of whom 37% had advanced lung cancer, 14% had breast cancer, 10% had colorectal cancer and 39% had other malignancies. Seventy genes were targeted and accuracy of ctDNA sequencing was assessed by comparing with matched tissue tests for 386 patients and frequencies of somatic ctDNA alterations per gene were compared to those previously described in tissue sequencing projects such as data from The Cancer Genome Atlas (TCGA).
It was noted that the ctDNA mutation patterns were highly concordant with tissue analysis as reported by the TCGA. The overall accuracy of ctDNA sequencing in comparison with matched tissue tests was 87% and the accuracy increased to 98% when blood and tumor were collected less than six months apart. Pearson Correlation between sets of data is a measure of how well these sets are related. Between 0.5 and 1.0 is considered high correlation. Pearson correlation for TP53 gene was 0.94, for KRAS was 0.99 and for PIK3CA was 0.99.
The researchers commented on the clinical outcome benefits using liquid biopsy, in four distinct groups:
1) Testing for actionable mutations (ALK fusion, EGFR or BRAF activating mutations in lung; ERBB2 amplification in gastric cancer) in cases with insufficient tissue quantity.
2) Testing for actionable resistance mutations (MET amplification or EGFR T790M in lung cancer), at the time of progression.
3) Genomic evolution upon progression such as ERBB2-amplified metastatic breast cancer in patients with triple negative primary tumor.
4) Tumors with genotypes that need more extensive driver mutation testing such as BRAF V600E in lung.
The authors concluded that there is a high correlation between ctDNA plasma samples and tissue testing with the exception of resistance mutations such as EGFR T790M mutation which evolve while on anti-EGFR inhibitor therapy and consequently may not correlate with the TCGA, probably because patients in the tissue-based population had not yet received the anti-EGFR inhibitor therapy that promotes the mutation. Patients who received treatment based on ctDNA findings also experienced better clinical outcomes. Zill OA, Mortimer S, Banks KC, et al Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. J Clin Oncol. 2016;34(suppl; abstr LBA11501).
Updated Survival Analysis of ONIVYDE®, 5-FU and Leucovorin Combination in Metastatic Pancreatic Carcinoma
SUMMARY: The American Cancer Society estimates that in 2016, over 53,000 people will be diagnosed with pancreatic cancer in the United States and close to 42,000 patients will die of the disease. Some important risk factors for Pancreatic cancer include increasing age, obesity, smoking history, genetic predisposition, exposure to certain dyes and chemicals, heavy alcohol use and pancreatitis. The best chance for long term survival is complete surgical resection, although this may not be feasible in a majority of the patients, as they present with advanced disease at the time of diagnosis. Based on the National Cancer Data Base, the 5 year observed survival rate for patients diagnosed with exocrine cancer of the Pancreas is 14% for those with Stage IA disease and 1% for those with Stage IV disease.
ONIVYDE® is a novel nanoliposomal encapsulation of Irinotecan, a topoisomerase 1 inhibitor. It is designed to optimize the delivery of Irinotecan, by extending the duration of circulation of the drug in the body and preferentially activating the drug within the tumor tissues, to achieve higher levels of the active cytotoxic drug metabolite, SN-38. This approach reduces the toxicity of Irinotecan to normal tissues while maintaining or increasing its anti-tumor efficacy.
NAPOLI-1 is an open-label phase III study in which 417 patients with Gemcitabine-refractory metastatic Pancreatic adenocarcinoma were randomly assigned in a 1:1:1 ratio to receive either ONIVYDE® monotherapy, ONIVYDE® plus 5-FluoroUracil (5-FU) and Leucovorin or 5-FU with Leucovorin (control group). Sixty one percent (61%) of patients had cancer in the head of the Pancreas and 68% had liver metastases. Treatment consisted of ONIVYDE® 120 mg/m2 IV over 90 minutes every 3 weeks in Group A, ONIVYDE® 80 mg/m2 IV given over 90 minutes followed by 5-FU 2400 mg/m2 given over 46 hours and racemic Leucovorin 400 mg/m2 IV given over 30 minutes every 2 weeks in Group B and 5-FU 2000 mg/m2 IV given over 24 hours plus racemic Leucovorin 200 mg/m2 IV given over 30 minutes weekly for 4 weeks followed by 2 weeks of rest in Group C (Control group). Each of the two ONIVYDE® containing groups was compared with the 5FU/Leucovorin control group. The primary study endpoint was Overall Survival and secondary endpoints included Progression Free Survival (PFS) and Overall Response Rate (ORR).
The primary survival analysis was previously reported was based on 313 events. The combination of ONIVYDE®, 5-FU and Leucovorin resulted in a median OS of 6.1 months compared with 4.2 months with 5-FU and Leucovorin alone (HR = 0.67; P=0.012). The median PFS was 3.1 months for the ONIVYDE® combination compared with 1.5 months with 5-FU and Leucovorin alone (HR= 0.55; P=0.0001). The ORR was low in both treatment groups (7.7% vs 0.8%), respectively. ONIVYDE® montherapy was not superior, compared with 5-FU and Leucovorin and was associated with more side effects compared to the combination regimen. In an expanded, pre-specified analyses, patients who received at least 80% of the target dose in the first 6 weeks experienced an even greater Overall Survival benefit (43% improvement) with ONIVYDE® combination, compared with 5-FU and Leucovorin alone (8.9 months vs 5.1 months, HR=0.57, P=0.011).
This publication is an updated analysis of OS along with 6 and 12 month survival estimates and safety, based on 378 events, as of 25 May 2015. The authors noted that that the combination of ONIVYDE®, 5-FU and Leucovorin maintained its median OS of 6.2 months compared with 4.2 months with 5-FU and Leucovorin alone (HR = 0.75; P=0.04). Again, there was no OS advantage with ONIVYDE® monotherapy, when compared with 5-FU and Leucovorin. The 6 month survival estimate was 53% with ONIVYDE®, 5-FU and Leucovorin compared with 38% for 5-FU and Leucovorin alone and 12 month survival estimates were 26% for ONIVYDE®, 5-FU and Leucovorin versus 16% for 5-FU and Leucovorin alone. The impact of ONIVYDE® on Overall Survival was more dramatic, with increasing benefit seen with higher levels of CA 19-9. The most common grade 3/4 adverse events with ONIVYDE® plus 5-FU and Leucovorin were neutropenia, fatigue, diarrhea and vomiting.
The authors following this updated analysis concluded that the median Overall Survival benefit for ONIVYDE® plus 5-FU and Leucovorin was maintained, with no new adverse events and this combination may be a new standard of care for patients with metastatic Pancreatic cancer, previously treated with Gemcitabine based therapy. Updated overall survival analysis of NAPOLI-1: Phase III study of nanoliposomal irinotecan (nal-IRI, MM-398), with or without 5-fluorouracil and leucovorin (5-FU/LV), versus 5-FU/LV in metastatic pancreatic cancer (mPAC) previously treated with gemcitabine-based therapy. Wang-Gillam A, Li C, Bodoky G, et al. J Clin Oncol 34, 2016 (suppl 4S; abstr 417)
First Oral Triplet Combination – NINLARO®, REVLIMID® and Dexamethasone for Multiple Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, about 30,330 new cases will be diagnosed in 2016 and 12,650 patients will die of the disease. Proteasomes are enzymes found in cells and they enable the breakdown of abnormal or mutant proteins. The amino acids from these proteins are recycled to make new proteins. Myeloma cells depend on the proteasomes to facilitate this metabolic function, to regulate their growth and survival. NINLARO® (Ixazomib) unlike VELCADE® (Bortezomib) is a second generation, oral, proteasome inhibitor, which disrupts protein metabolism in Myeloma cells, by inhibiting proteasomes and has an antiproliferative and pro-apoptotic effect.
The approval of NINLARO® was based a pivotal, multicenter, randomized, double-blind, placebo-controlled, phase III trial (TOURMALINE-MM1 study), in which 722 patients with Multiple Myeloma were randomized in a 1:1 ratio to receive either a combination of NINLARO®, REVLIMID® and Dexamethasone (N=360) or a combination of Placebo, REVLIMID® and Dexamethasone (n=362). NINLARO® was administered at 4 mg PO on days 1, 8, and 15 in combination with REVLIMID® 25 mg PO on days 1 thru 21 and Dexamethasone 40 mg PO on days 1, 8, 15, and 22 of a 28 day treatment cycle. Treatment was continued until disease progression or unacceptable toxicity. Enrolled patients had received 1 to 3 prior lines of therapy, which included VELCADE® (69%), THALOMID® (45%), and REVLIMID® (12%) and 77% of the patients had relapsed Multiple Myeloma. The median age of patients was 66 years. The primary end point of the study was Progression Free survival (PFS) and secondary endpoints included Objective Response Rate (ORR), safety, and Overall Survival.
At a median follow-up of 14.7 months, the PFS with the combination of NINLARO®, REVLIMID® and Dexamethasone was 20.6 months compared with 14.7 months for the combination group of Placebo, REVLIMID® and Dexamethasone (HR= 0.74, P=0.01). This benefit in the NINLARO® group, was noted in all prespecified patient subgroups, including those with high risk cytogenetic abnormalities. The Objective Response Rate was 78% in the NINLARO® group and 72% in the placebo group, and the Complete Response plus Very Good Partial Response in these two treatment groups were 48% and 39% respectively. The median time to response was 1.1 months in the NINLARO® group and 1.9 months in the placebo group and the median duration of response was 20.5 months and 15.0 months respectively. At a median follow up of 23 months, the Overall Survival has not been reached in either study group. Serious adverse events (at least grade 3) were similar in the two study groups (47% in the NINLARO® group and 49% in the placebo group). Patients in the NINLARO® group experienced more adverse events such as thrombocytopenia, vomiting, diarrhea, peripheral neuropathy and skin rash. However, patient-reported Quality of Life was similar in both treatment groups.
The authors concluded that NINLARO® based oral triplet therapy significantly prolonged Progression Free Survival compared with REVLIMID® and Dexamethasone, in patients with relapsed/refractory Multiple Myeloma, with acceptable toxicities. Studies are underway, evaluating NINLARO® in newly diagnosed Myeloma patients as well as maintenance therapy in non-transplant patients. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. Moreau P, Masszi T, Grzasko N, et al. N Engl J Med 2016; 374:1621-1634
Recurrent VTE in Cancer Patients Treated with XARELTO®
SUMMARY: The Center for Disease Control and Prevention (CDC) estimates that approximately 1-2 per 1000 individuals develop Deep Vein Thrombosis/Pulmonary Embolism (PE) each year in the United States, resulting in 60,000 – 100,000 deaths. Venous ThromboEmbolism (VTE) is the third leading cause of cardiovascular mortality. Patients with unprovoked DVT and PE are two to four times more likely to be diagnosed with cancer within the following 12 months compared to the general population. In patients with cancer associated thrombosis, COUMADIN® (Warfarin) and XARELTO® (Rivaroxaban) are often prescribed, despite guidelines recommending Low Molecular Weight Heparin (LMWH) in this patient population.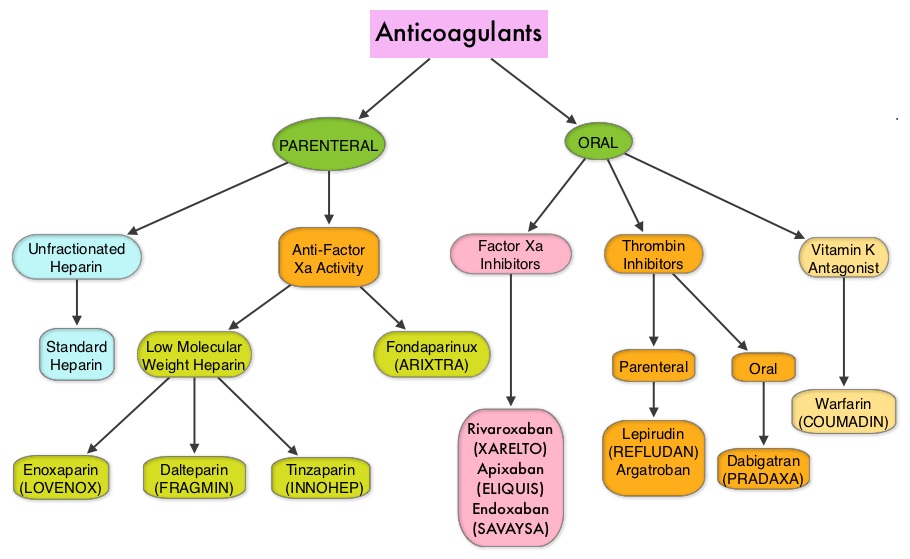
Recently published data suggests that the rates of major bleeding, with use of XARELTO® in a highly selected group of cancer patients with venous thromboembolic disease, compared favorably with those treated with LMWH. (Mantha S, et al. 2015 ASH Annual Meeting). There is however limited data comparing the efficacy of different anticoagulants for VTE treatment in cancer patients.
The authors conducted this study in cancer patients, to compare the VTE recurrence rates, following most frequently prescribed anticoagulants in the United States. Newly diagnosed cancer patients with a first VTE, who initiated LMWH, COUMADIN® or XARELTO®, were selected using healthcare claims from the Humana database. The study population included 2,428 patients (XARELTO®: N=707; LMWH: N=660; COUMADIN® N =1,061). VTE recurrences were defined as hospitalizations with a primary diagnosis of VTE. Outpatients with a primary diagnosis of VTE were added as a sensitivity analysis to the recurrence definition.
The median duration on initial LMWH treatment was 1 month, on COUMADIN® was 3.5 months and on XARELTO® was 3 months. When compared to LMWH, VTE recurrence rates were lower with initial XARELTO® treatment at 6 months (13.2% versus 17.1%; P=0.06) and at 12 months (16.5% versus 22.2%; P=0.03). When initially treated with XARELTO®, recurrent VTE was 28% less likely than with LMWH (HR=0.72; P<0.03).
When compared to COUMADIN®, VTE recurrence rates were again lower with initial XARELTO® treatment at 6 months (13.2% versus 17.5%; P=0.02) and at 12 months (15.7% versus 19.9%; P=0.02). When initially treated with XARELTO®, recurrent VTE was 26% less likely than with COUMADIN® (HR=0.74; P<0.03). This benefit with XARELTO® when compared with LMWH and COUMADIN® users, was also noted in the sensitivity analysis.
The authors concluded that based on this real world healthcare claims data in cancer patients, XARELTO® was associated with a lower risk of recurrent VTE than LMWH or COUMADIN® and this could be a reflection of a shorter duration of treatment with LMWH and difficult therapeutic anticoagulation with COUMADIN®. Recurrent VTE in cancer patients treated with anticoagulation. Streiff MB, Milentijevic D, McCrae K, et al. J Clin Oncol 34, 2016 (suppl; abstr 10024)
Daily Aspirin May Improve Survival after Diagnosis of Colorectal Cancer
SUMMARY: ColoRectal Cancer (CRC) is the third most common cancer diagnosed in both men and women in the United States. The American Cancer Society estimates that approximately 135,000 new cases of ColoRectal Cancer will be diagnosed in the United States in 2016 and over 49,000 patients are expected to die of the disease. Several epidemiological studies as well as randomized controlled trials have shown that Aspirin and NonSteroidal Anti-Inflammatory Drugs (NSAIDs) reduce the incidence of ColoRectal Cancer (CRC) and CRC associated mortality. Nonetheless, use of aspirin for the primary prevention of CRC, is not routinely recommended, for the fear of aspirin-induced gastric and cerebral hemorrhages. Even though the benefits of Aspirin in the primary prevention of CRC remains well established, the role of Aspirin in secondary prevention in patients with CRC (after the diagnosis of CRC) is unclear. Platelets have long been implicated in the mechanism of tumor metastases. More recent data suggests that platelets may play a role in tumorigenesis as well, through the release of angiogenic and growth factors due to overexpression of COX-2. Daily low dose Aspirin inhibits COX-1 and COX-2. It is postulated that Aspirin also works by COX-independent mechanisms such as, the inhibition of NF-kB and Wnt/ β-catenin signaling, which may play a role in its chemopreventive properties.
Even though the benefits of Aspirin in the primary prevention of CRC remains well established, the role of Aspirin in secondary prevention in patients with CRC (after the diagnosis of CRC) is unclear. Platelets have long been implicated in the mechanism of tumor metastases. More recent data suggests that platelets may play a role in tumorigenesis as well, through the release of angiogenic and growth factors due to overexpression of COX-2. Daily low dose Aspirin inhibits COX-1 and COX-2. It is postulated that Aspirin also works by COX-independent mechanisms such as, the inhibition of NF-kB and Wnt/ β-catenin signaling, which may play a role in its chemopreventive properties.
The authors conducted this trial to evaluate the association between Aspirin use after diagnosis of CRC and CRC-Specific Survival (CSS) and Overall Survival (OS), in the largest group of patients ever studied. The study authors in this retrospective, population-based study identified 24,495 patients in the Cancer Registry of Norway, diagnosed with ColoRectal Cancer from 2004 through 2011 and a total of 23,162 patients diagnosed with CRC were included. Using the Norwegian Prescription Database, the authors were then able to establish that 6,102 patients in this large cohort had documented exposure to Aspirin. Exposure to Aspirin was defined as a prescription for more than 6 months of Aspirin, following a diagnosis of CRC. The median follow up was 3 years.
The authors performed a multivariate regression analysis controlling for age, gender, tumor stage, tumor differentiation and noted that exposure to Aspirin post-diagnosis, independently improved ColoRectal Cancer-Specific Survival (HR=0.85; P<0.001) and Overall Survival (HR=0.95; P<0.076). Patients who used Aspirin both before and after diagnosis of CRC had additional improvement in ColoRectal Cancer-Specific Survival (HR= 0.77; P<0.001) and Overall Survival (HR=0.86; P<0.001). Further, patients with poorly and moderately differentiated tumors experienced the greatest benefits of Aspirin exposure, as well as those with stage II disease. The researchers were also able to demonstrate that Aspirin use was most beneficial in the first 2 to 3 years after diagnosis.
It was concluded that Aspirin use after the diagnosis of ColoRectal Cancer, is independently associated with improved Colorectal Cancer-Specific Survival and Overall Survival. Because of the increased risk for bleeding, the risk-benefit should be assessed before Aspirin is routinely recommended to this patient population. Aspirin as Secondary Prevention in Patients With Colorectal Cancer: An Unselected Population-Based Study. Bains SJ, Mahic M, Myklebust TA, et al. Published online before print May 31, 2016, doi:10.1200/JCO.2015.65.3519JCO May 31, 2016 JCO653519
Late Breaking Abstract – ASCO 2016 Extended Adjuvant AI Therapy Improves DFS in Postmenopausal Hormone Receptor Positive Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 246,660 new cases of invasive breast cancer will be diagnosed in 2016 and 40,450 women will die of the disease. Approximately 75% of patients with breast cancer are hormone receptor positive (Estrogen Receptor/Progesterone Receptor positive) and this is a predictor of response to endocrine therapy. These patients are often treated with anti-estrogen therapy as first line treatment. In premenopausal woman, the ovary is the main source of estrogen production, whereas in postmenopausal women, the primary source of estrogen is the Aromatase enzyme mediated conversion of androstenedione and testosterone to estrone and estradiol, in extragonadal/peripheral tissues. NOLVADEX® (Tamoxifen) is a nonsteroidal Selective Estrogen Receptor Modulator (SERM) and works mainly by binding to the Estrogen Receptor and thus blocks the proliferative actions of estrogen on the mammary tissue. ARIMIDEX® (Anastrozole), FEMARA® (Letrozole) and AROMASIN® (Exemestane) are Aromatase Inhibitors (AIs) that binds to the Aromatase enzyme and inhibit the conversion of androgens to estrogens in the extra-gonadal tissues. Postmenopausal women with hormone receptor positive early breast cancer are often treated with 5 years of Aromatase Inhibitor (AI) therapy either as up-front treatment or after 2-5 years of Tamoxifen. The benefit of extending treatment with an AI to 10 years may further reduce the risk of breast cancer recurrence, but this benefit was not previously known.
The Canadian Cancer Trials Group MA.17R is a double blind, placebo controlled, phase III trial, which tested the benefit of extending AI treatment, using FEMARA®, for an additional 5 years. This study involved 1,918 postmenopausal women with early stage breast cancer and included three patient groups – one group had no prior treatment with adjuvant Tamoxifen whereas the other two groups had adjuvant Tamoxifen for some duration of time. All patients however had recently received 4.5 to 6 years of adjuvant AI therapy. These patients were then randomly assigned to receive either extended adjuvant treatment with FEMARA® or placebo for an additional five years. The primary endpoint was Disease Free Survival (DFS).
After a median follow up of 6.3 years, the 5 year DFS rate for the extended FEMARA® group was 95% compared with 91% for the placebo group (HR=0.66; P=0.01). The improvement in DFS was significant among patients with node-positive disease, but not for those with node-negative disease. There was no difference in the 5 year Overall Survival between the two groups – 93% with FEMARA® versus 94% with placebo (HR 0.97; P=NS). The annual incidence rate of contralateral breast cancer was however significantly better in the FEMARA® group at 0.21%, compared with 0.49% with placebo (HR=0.42; P=0.007). Patients receiving extended treatment with FEMARA® had more frequent adverse events such as bone pain, elevation of alkaline phosphatase, and elevation of alanine transaminase. There was also a greater incidence of osteoporosis with FEMARA® than with placebo (11% vs 6%; P<0.0001) and fracture risk was higher in the FEMARA® group (14%) compared with 9% in the placebo group (P=0.001).
The authors concluded that this is the first study to show added benefit of improved Disease Free Survival, by extending an adjuvant AI beyond 5 years to 10 years, when compared with 5 years of AI treatment as initial therapy or preceded by 2-5 years of Tamoxifen. A randomized trial (MA.17R) of extending adjuvant letrozole for 5 years after completing an initial 5 years of aromatase inhibitor therapy alone or preceded by tamoxifen in postmenopausal women with early-stage breast cancer. Goss PE, Ingle JN, Pritchard KI, et al. J Clin Oncol 34, 2016 (suppl; abstr LBA1)