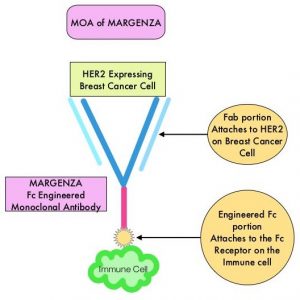
SUMMARY: It is estimated that in the United States, approximately 21,750 women will be diagnosed with ovarian cancer in 2020 and 13,940 women will die of the disease. Ovarian cancer ranks fifth in cancer deaths among women, and accounts for more deaths than any other cancer of the female reproductive system. Approximately 75% of the ovarian cancer patients are diagnosed with advanced disease. Approximately 85% of all ovarian cancers are epithelial in origin, and approximately 70% of all epithelial ovarian cancers are High-Grade Serous adenocarcinomas. Patients with newly diagnosed advanced ovarian cancer are often treated with platinum based chemotherapy following primary surgical cytoreduction. Approximately 70% of these patients will relapse within the subsequent 3 years and are incurable, with a 5 year Overall Survival rate of about 20-30%.
Germline mutations in BRCA1 and BRCA2 genes account for about 17% of ovarian cancers (mutations present in all individual cells), whereas somatic mutations are found in an additional 7% (mutations present exclusively in tumor cells). BRCA1 and BRCA2 are tumor suppressor genes and they recognize and repair double strand DNA breaks via Homologous Recombination (HR) pathway. Homologous Recombination is a DNA repair pathway utilized by cells to accurately repair DNA double-stranded breaks during the S and G2 phases of the cell cycle, and thereby maintain genomic integrity. The PARP (Poly ADP Ribose Polymerase) family of enzymes include PARP1 and PARP2, and is a related enzymatic pathway that repairs single strand breaks in DNA. In a BRCA mutant, the cancer cell relies solely on PARP pathway for DNA repair to survive. PARP inhibitors traps PARP onto DNA at sites of single-strand breaks, thereby preventing their repair and generate double-strand breaks. These breaks cannot be repaired accurately in tumors harboring defects in Homologous Recombination Repair pathway genes, such as BRCA1 or BRCA2 mutations, and this leads to cumulative DNA damage and tumor cell death.
This systematic review-based guideline was developed by a multidisciplinary ASCO Expert Panel to provide clinicians and other health care practitioners, recommendations on the use of PARP inhibitors for management of Epithelial Ovarian, tubal, or Primary Peritoneal Cancer (herein referred to as EOC), based on best available evidence. The recommendations were developed following a systematic review of the literature which identified 17 randomized controlled trials published from 2011 through 2020, that included patients who have not previously received a PARP inhibitor.
ASCO Guideline Questions:
1) Should PARP inhibitor therapy for EOC be repeated over the course of treatment?
2) In which patients with newly diagnosed EOC are PARP inhibitors recommended?
a. What are the histologic types of EOC for which PARP inhibitors are recommended?
b. What are the biomarker subsets for which PARP inhibitors are recommended?
3) Is PARP inhibitor monotherapy recommended for recurrent EOC? If so,
a. In which settings (eg, second-line maintenance or treatment of recurrent disease)?
b. At what dose and duration?
4) Are there settings where PARP inhibitors in combination with chemotherapy or other targeted therapy are recommended?
5) How should clinicians manage the specific toxicities of the various PARP inhibitors?
Recommendations: The following recommendations pertain only to patients with EOC who have not previously received a PARP inhibitor.
Repeating PARP Inhibitor
Recommendation 1.0: Repeating therapy with a PARP inhibitor in the treatment of EOC is not recommended at this time. Consideration should be made as to the best time in the life cycle of an individual patient’s EOC in which to use PARP inhibitor. Clinical trial participation is encouraged.
Newly Diagnosed Ovarian Cancer
Recommendation 2.0: PARP inhibitors are not recommended for use in initial treatment of early stage (Stage I-II) EOC because there is insufficient evidence to support use in this population.
Recommendation 2.1: Women with newly diagnosed Stage III-IV EOC that is in Complete or Partial Response to first-line platinum-based chemotherapy should be offered PARP inhibitor maintenance therapy with Olaparib (for those with germline or somatic pathogenic or likely pathogenic variants in BRCA1 or BRCA2 genes) or Niraparib (all women) in High-Grade Serous or endometrioid ovarian cancer.
PARP inhibitor maintenance therapy should consist of Olaparib (300 mg orally every 12 hours for 2 years) or Niraparib (200-300 mg orally daily for 3 years). Longer duration could be considered in selected individuals.
Recommendation 2.2: The addition of Olaparib to Bevacizumab maintenance may be offered to patients who have Stage III-IV High-Grade Serous or endometrioid ovarian cancer and germline or somatic pathogenic or likely pathogenic variants in BRCA1 or BRCA2 genes and/or genomic instability, as determined by Myriad myChoice CDx, and who have had a Partial or Complete Response to chemotherapy plus Bevacizumab combination.
Recommendation 2.3: Inclusion of the PARP inhibitor Veliparib with combination chemotherapy followed by Veliparib maintenance therapy cannot be recommended at this time. There are no data that this approach is superior, equal, or less toxic than a switch maintenance.
Note: Veliparib is not commercially available at the time of these recommendations.
Recurrent Ovarian Cancer: Second-Line or Greater Maintenance and Treatment
Recommendation 3.0: PARP inhibitor monotherapy maintenance (second-line or more) may be offered to patients with EOC who have not already received a PARP inhibitor and who have responded to platinum-based therapy regardless of BRCA mutation status. Treatment is continued until disease progression or toxicity despite dose reductions and best supportive care. Options include Olaparib 300 mg every 12 hours, Rucaparib 600 mg every 12 hours or Niraparib 200-300 mg once daily.
Recommendation 3.1: Treatment with a PARP inhibitor should be offered to patients with recurrent EOC who have not already received a PARP inhibitor and have a germline or somatic pathogenic or likely pathogenic variants in BRCA1 or BRCA2 genes. Options include Olaparib 300 mg every 12 hours, Rucaparib 600 mg every 12 hours or Niraparib 200-300 mg once daily.
Recommendation 3.2: Treatment with a PARP inhibitor monotherapy should be offered to patients with recurrent EOC who have not already received a PARP inhibitor, and whose tumor demonstrates genomic instability, as determined by Myriad myChoice CDx, and has not recurred within 6 months of platinum-based therapy
Recommendation 3.3: PARP inhibitors are not recommended for treatment of BRCA wild-type or platinum-resistant recurrent EOC
PARP Inhibitors in Combination
Recommendation 4.0: PARP inhibitors are not recommended for use in combination with chemotherapy, other targeted agents, or immune-oncology agents in the recurrent setting outside the context of a clinical trial. Clinical trial participation is encouraged.
Management of Adverse Events
Recommendation 5.0 Anemia: Patients requiring a blood transfusion for symptom relief and/or hemoglobin level less than 8 g/dL should be monitored. PARP inhibitor dose should be reduced with evidence of repeated anemia to avoid multiple transfusions. Patients with progressive anemia may be offered growth factor per ASCO guidelines and physician and patient comfort.
Recommendation 5.1 Neutropenia: Growth factor is not indicated for use in patients receiving daily PARP inhibitor. Neutropenia (grade 4 lasting at least 5-7 days or associated with fever) should result in dose hold until recovery of infection and granulocyte count, followed by dose reduction. Growth factor support may be used in this setting to support patient safety during the drug hold period.
Recommendation 5.2 Platelets: Thrombocytopenia is most common with Niraparib. Niraparib dosing guidelines should be used to lower starting dose (200 mg) based on weight and platelet count. Discontinue PARP inhibitor for persistent thrombocytopenia or significant bleeding despite dose reduction.
Recommendation 5.3 Persistent cytopenia: Evaluation for treatment-related Myelodysplastic Syndrome/Acute Myeloid Leukemia should be initiated in patients with persistent cytopenia that occurs despite drug hold.
Recommendation 5.4 Nausea: Many patients will have tachyphylaxis of nausea symptoms over the first cycle of therapy. Persistent nausea requiring daily antiemetic intervention, causing a reduction in performance status, and/or resulting in more than 5% weight loss, should result in dose reduction.
PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. Tew WP, Lacchetti C, Ellis A, et al. J Clin Oncol 2020;38:3468-3493.