
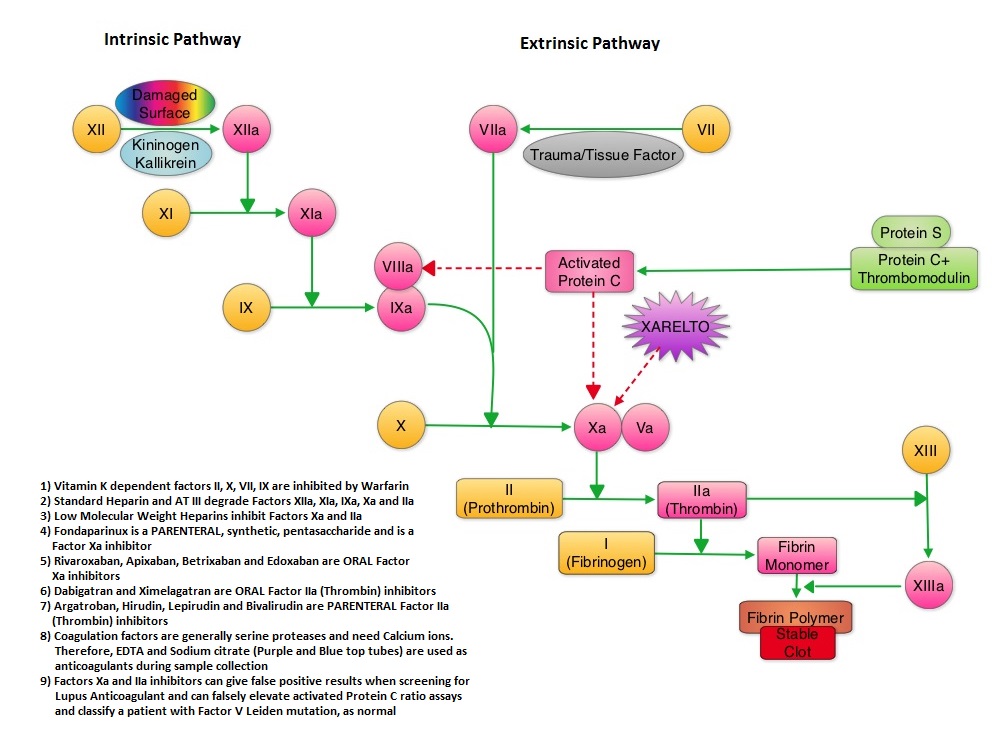
SUMMARY: XARELTO® (Rivaroxaban) is an oral, direct inhibitor of Factor Xa. Two previously published randomized studies concluded that XARELTO® was noninferior to the standard anticoagulation therapy (administration of heparin overlapped and followed by Vitamin K Antagonist), for most patients with Venous ThromboEmbolism. Further, XARELTO® in these studies also had a better safety profile compared to standard anticoagulant therapy. The authors in this pooled analysis combined the data from 2 identically designed studies, EINSTEIN-DVT and EINSTEIN-PE in which XARELTO® was compared to standard anticoagulation therapy, in patients with DVT and PE respectively. The goal of this study was to provide a more accurate estimate of the efficacy and safety of XARELTO® in elderly patients and cancer patients in whom Vitamin K Antagonists (VKA) such as COUMADIN® (Warfarin) can be associated with a higher complication rate. In addition, the study included analysis of outcomes with XARELTO® in patients with previous VTE and in those presenting with extensive thrombosis. Of the 8282 patients with VTE randomized in the pooled analysis, 4151 patients received XARELTO® and 4131 patients received LOVENOX® (Enoxaparin) / Vitamin K Antagonists. In both pooled studies, patients in the XARELTO® group received a dose of 15 mg PO BID for 21 days, followed by 20 mg PO QD whereas patients assigned to the standard anticoagulation therapy group received LOVENOX® subcutaneous at a dose of 1.0 mg/kg BID and either oral Warfarin or Acenocoumarol started within 48 hours after randomization and patients continued treatment for 3, 6, or 12 months, as determined by the local Health Care Providers. INR was closely monitored and maintained between 2-3. On final review, the analysis suggested that XARELTO® resulted in efficacy similar to standard anticoagulation therapy, with a noninferiority P<0.001. The pre-specified principal safety outcome was clinically relevant bleeding and major bleeding was observed in 40 patients belonging to the XARELTO® group and in 72 patients belonging to the standard anticoagulation therapy group (HR=0.54; P= 0.002). Similar benefits in the efficacy and safety were seen in the key subgroups evaluated, which included elderly fragile patients, cancer patients, patients presenting with extensive thrombosis and those with a history of recurrent VTE. The authors concluded that the incidence of major bleeding with XARELTO® was significantly less, particularly in the high risk group of patients, when compared to standard anticoagulation therapy and may therefore have a safety advantage, without compromising efficacy. Prins MH, Lensing AW, Bauersachs R, et al. Thrombosis Journal 2013;11:21
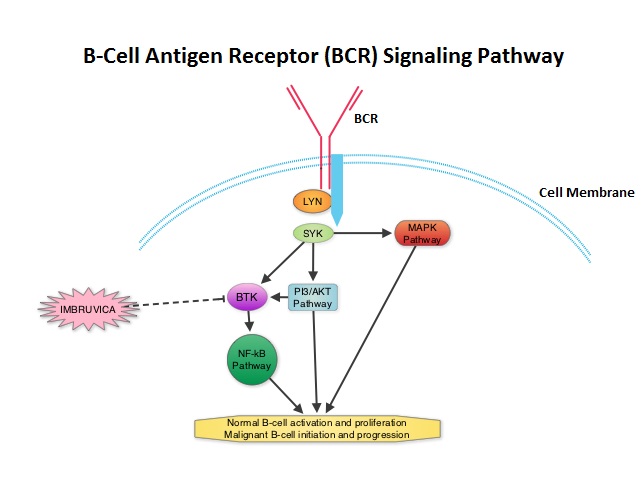 The FDA granted accelerated approval of IMBRUVICA® for the treatment of patients with Chronic Lymphocytic Leukemia (CLL) who had received at least one prior therapy. This approval was based on the outcomes in a select group of 48 patients who were a part of a larger group of 85 patients, enrolled in a multicenter single arm phase Ib/II trial. The median age was 67 years and 71% were male. Patients had a median number of 4 prior treatments and had an ECOG PS of 0-1. Patients in this group received IMBRUVICA® 420 mg PO daily until disease progression or unacceptable toxicity. The overall response rate was 58.3% as assessed by an independent review committee. No complete responses were seen and the response duration ranged from 5.6 to over 24 months. This analysis did not include data from those patients enrolled in the trial who received IMBRUVICA® 840 mg PO daily or those with Small Lymphocytic Lymphoma (N=37). The most common toxicities included fatigue, myalgias and arthralgias, cytopenias, nausea, diarrhea, fever and rash. Transient asymptomatic increase in lymphocyte count with resolution of lymphadenopathy and splenomegaly was common but resolved with continued treatment. The confirmatory RESONATE trial is a multicenter, randomized, open-label Phase III study in which single agent IMBRUVICA® was compared to single agent ARZERRA® (Ofatumumab) in patients with relapsed or refractory CLL or Small Lymphocytic Lymphoma . This was a part of the requirement by the FDA. Enrolled patients had measurable nodal disease and were not eligible for treatment with purine analog-based therapy. In this study, 391 patients who had received at least one prior therapy, were enrolled and randomized to receive 420 mg of IMBRUVICA® orally once daily or ARZERRA® given intravenously. Treatment was given over a period of 24 weeks until disease progression or unacceptable toxicity. Patients randomized to the ARZERRA® group on disease progression were allowed to receive treatment with IMBRUVICA®. The primary endpoint of this study was progression-free survival and the secondary endpoint was overall survival. Following recommendations from the Independent Data Monitoring Committee (IDMC), the study was stopped earlier, as the primary endpoint as well as an important secondary endpoint of the study were met. At the planned interim analysis, patients in the IMBRUVICA® group showed a statistically significant improvement in progression-free survival, the primary endpoint of the study as well as a statistically significant improvement in overall survival, the secondary endpoint of the trial. This data confirmed the efficacy of IMBRUVICA® and gives patients with CLL, an important new treatment option. Byrd JC, Furman RR, Coutre SE, et al. N Engl J Med 2013; 369:32-42
The FDA granted accelerated approval of IMBRUVICA® for the treatment of patients with Chronic Lymphocytic Leukemia (CLL) who had received at least one prior therapy. This approval was based on the outcomes in a select group of 48 patients who were a part of a larger group of 85 patients, enrolled in a multicenter single arm phase Ib/II trial. The median age was 67 years and 71% were male. Patients had a median number of 4 prior treatments and had an ECOG PS of 0-1. Patients in this group received IMBRUVICA® 420 mg PO daily until disease progression or unacceptable toxicity. The overall response rate was 58.3% as assessed by an independent review committee. No complete responses were seen and the response duration ranged from 5.6 to over 24 months. This analysis did not include data from those patients enrolled in the trial who received IMBRUVICA® 840 mg PO daily or those with Small Lymphocytic Lymphoma (N=37). The most common toxicities included fatigue, myalgias and arthralgias, cytopenias, nausea, diarrhea, fever and rash. Transient asymptomatic increase in lymphocyte count with resolution of lymphadenopathy and splenomegaly was common but resolved with continued treatment. The confirmatory RESONATE trial is a multicenter, randomized, open-label Phase III study in which single agent IMBRUVICA® was compared to single agent ARZERRA® (Ofatumumab) in patients with relapsed or refractory CLL or Small Lymphocytic Lymphoma . This was a part of the requirement by the FDA. Enrolled patients had measurable nodal disease and were not eligible for treatment with purine analog-based therapy. In this study, 391 patients who had received at least one prior therapy, were enrolled and randomized to receive 420 mg of IMBRUVICA® orally once daily or ARZERRA® given intravenously. Treatment was given over a period of 24 weeks until disease progression or unacceptable toxicity. Patients randomized to the ARZERRA® group on disease progression were allowed to receive treatment with IMBRUVICA®. The primary endpoint of this study was progression-free survival and the secondary endpoint was overall survival. Following recommendations from the Independent Data Monitoring Committee (IDMC), the study was stopped earlier, as the primary endpoint as well as an important secondary endpoint of the study were met. At the planned interim analysis, patients in the IMBRUVICA® group showed a statistically significant improvement in progression-free survival, the primary endpoint of the study as well as a statistically significant improvement in overall survival, the secondary endpoint of the trial. This data confirmed the efficacy of IMBRUVICA® and gives patients with CLL, an important new treatment option. Byrd JC, Furman RR, Coutre SE, et al. N Engl J Med 2013; 369:32-42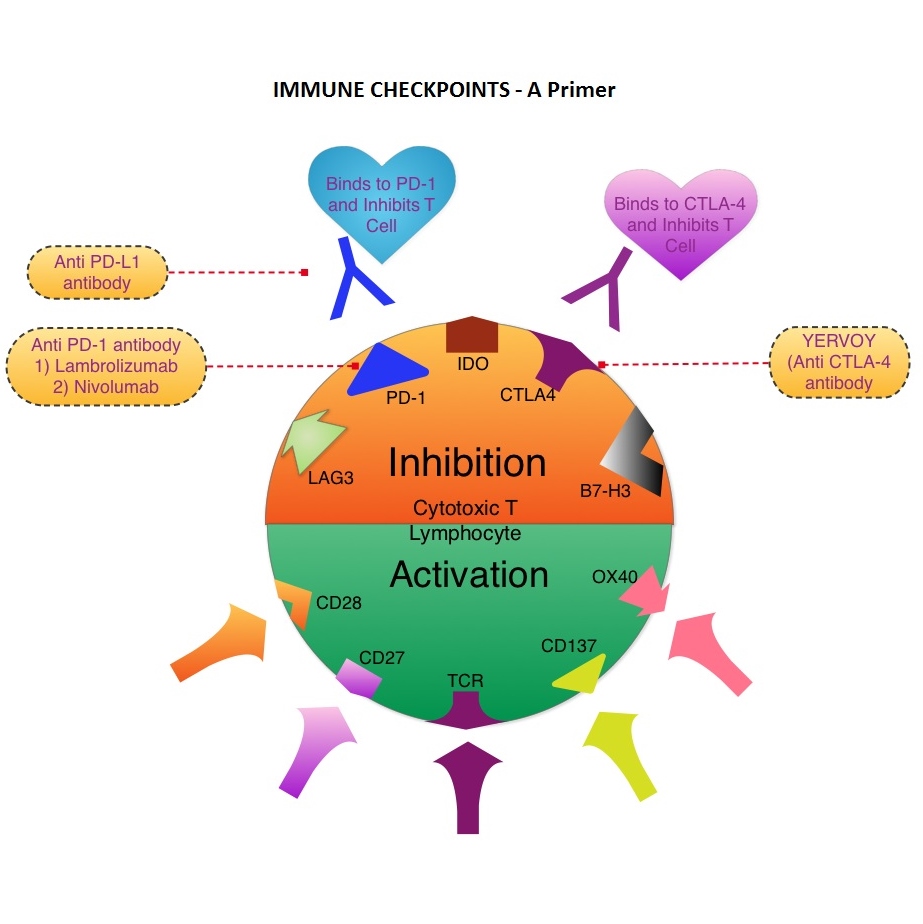 The T cells of the immune system play a very important role in modulating the immune system. EFFECTOR T cells include Cytotoxic T cells, Helper T cells, and Natural Killer (NK) cells, that enable the immune system to destroy cancer cells and pathogens. The REGULATORY T cells however, suppress immune response. Under normal circumstances, inhibition of an intense immune response and switching off the EFFECTOR T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. The mechanism can be compared to a lock and key where the appropriate Ligand (KEY) binds to the Immune checkpoint protein/receptor (LOCK) and activates or inhibits a T lymphocyte. With the ongoing understanding of tumor immunology and the recognition of Immune checkpoint proteins, researchers have focused on the development of antibodies that either target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4, PD-1, IDO, etc. (LOCK) or target the inhibitory soluble Ligands or antigens that are located on the surface of certain cancer cells (KEY) that bind to these Immune check point proteins/receptors. By doing so, one would expect to unleash the EFFECTOR T cells resulting in T cell proliferation, activation and a therapeutic response. The first immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® (Ipilimumab), an antibody that blocks Immune checkpoint protein/receptor CTLA-4, was approved by the FDA in March 2011 and has been shown to prolong overall survival in patients with previously treated unresectable or metastatic melanoma. The next immune check point protein/receptor studied for targeted therapy was PD-1. Lambrolizumab (MK-3475) is a humanized anti–PD-1 monoclonal antibody that demonstrated a 38% rapid and durable response rate and a more than 7 month median progression-free survival in patients with advanced melanoma, regardless of their prior therapy with YERVOY®. Nivolumab, another PD-1 targeted antibody demonstrated remarkable efficacy in a Phase I study with an overall response rate of 30%, median survival of 16.8 months and a 2 year survival of 44%. Based on this provocative data, a combination of Nivolumab and YERVOY® were studied in patients with advanced Stage III or IV melanoma who had received up to three prior therapies.. The idea was to block both the Immune checkpoints, PD-1 and CTLA-4, for improved efficacy. Fifty three (N=53) patients were treated with a combination of these two agents and 33 patients received these agents sequentially. Indeed, the highest response rate was over 50% in the combination group with 30% of these patients experienced a more than 80% response rate at 12 weeks of treatment whereas the response rate in the sequential treatment group was 20%. This preliminary study confirmed that blocking multiple Immune checkpoint proteins/receptors may result in rapid and durable responses in patients with advanced malignant melanoma. Phase III studies are underway to confirm this efficacy data and this concept is also being studied in other tumor types. Targeting/inhibiting the ligands (KEY) and preventing their binding to the Immune checkpoint protein/receptor, is another approach to stimulate antitumor immune response. PD-L1 protein (Ligand) which is often elevated in melanoma tumor cells, bind to PD-1 check point protein/receptor and can inhibit T cells and escape immune surveillance. An investigational PD-L1 targeted (Ligand targeted) engineered antibody (MPDL3280A) demonstrated a rapid response in 26% of the 45 patients with metastatic melanoma and the benefit was more so in those tumors expressing PD-L1. Promising activity has also been seen in advanced renal cell carcinoma. Antibodies targeting the Immune checkpoint receptor/protein or the Ligands binding to these receptors, are being developed, to carry payloads that are lethal to the checkpoint protein/receptor or Ligand. In conclusion, identifying as well as inhibiting certain Immune checkpoint proteins/receptors and/or Ligands that bind to these receptors, may give us new insights in the field of tumor immunology, resulting in better outcome for our cancer patients. Patel JD, Krilov L, Adams S, et al. J Clin Oncol 2013;32:129-160
The T cells of the immune system play a very important role in modulating the immune system. EFFECTOR T cells include Cytotoxic T cells, Helper T cells, and Natural Killer (NK) cells, that enable the immune system to destroy cancer cells and pathogens. The REGULATORY T cells however, suppress immune response. Under normal circumstances, inhibition of an intense immune response and switching off the EFFECTOR T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. The mechanism can be compared to a lock and key where the appropriate Ligand (KEY) binds to the Immune checkpoint protein/receptor (LOCK) and activates or inhibits a T lymphocyte. With the ongoing understanding of tumor immunology and the recognition of Immune checkpoint proteins, researchers have focused on the development of antibodies that either target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4, PD-1, IDO, etc. (LOCK) or target the inhibitory soluble Ligands or antigens that are located on the surface of certain cancer cells (KEY) that bind to these Immune check point proteins/receptors. By doing so, one would expect to unleash the EFFECTOR T cells resulting in T cell proliferation, activation and a therapeutic response. The first immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® (Ipilimumab), an antibody that blocks Immune checkpoint protein/receptor CTLA-4, was approved by the FDA in March 2011 and has been shown to prolong overall survival in patients with previously treated unresectable or metastatic melanoma. The next immune check point protein/receptor studied for targeted therapy was PD-1. Lambrolizumab (MK-3475) is a humanized anti–PD-1 monoclonal antibody that demonstrated a 38% rapid and durable response rate and a more than 7 month median progression-free survival in patients with advanced melanoma, regardless of their prior therapy with YERVOY®. Nivolumab, another PD-1 targeted antibody demonstrated remarkable efficacy in a Phase I study with an overall response rate of 30%, median survival of 16.8 months and a 2 year survival of 44%. Based on this provocative data, a combination of Nivolumab and YERVOY® were studied in patients with advanced Stage III or IV melanoma who had received up to three prior therapies.. The idea was to block both the Immune checkpoints, PD-1 and CTLA-4, for improved efficacy. Fifty three (N=53) patients were treated with a combination of these two agents and 33 patients received these agents sequentially. Indeed, the highest response rate was over 50% in the combination group with 30% of these patients experienced a more than 80% response rate at 12 weeks of treatment whereas the response rate in the sequential treatment group was 20%. This preliminary study confirmed that blocking multiple Immune checkpoint proteins/receptors may result in rapid and durable responses in patients with advanced malignant melanoma. Phase III studies are underway to confirm this efficacy data and this concept is also being studied in other tumor types. Targeting/inhibiting the ligands (KEY) and preventing their binding to the Immune checkpoint protein/receptor, is another approach to stimulate antitumor immune response. PD-L1 protein (Ligand) which is often elevated in melanoma tumor cells, bind to PD-1 check point protein/receptor and can inhibit T cells and escape immune surveillance. An investigational PD-L1 targeted (Ligand targeted) engineered antibody (MPDL3280A) demonstrated a rapid response in 26% of the 45 patients with metastatic melanoma and the benefit was more so in those tumors expressing PD-L1. Promising activity has also been seen in advanced renal cell carcinoma. Antibodies targeting the Immune checkpoint receptor/protein or the Ligands binding to these receptors, are being developed, to carry payloads that are lethal to the checkpoint protein/receptor or Ligand. In conclusion, identifying as well as inhibiting certain Immune checkpoint proteins/receptors and/or Ligands that bind to these receptors, may give us new insights in the field of tumor immunology, resulting in better outcome for our cancer patients. Patel JD, Krilov L, Adams S, et al. J Clin Oncol 2013;32:129-160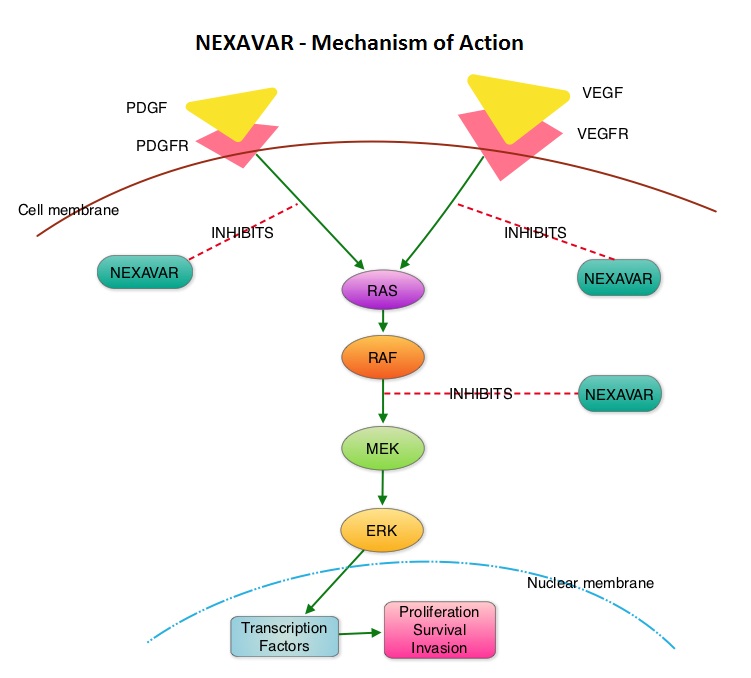 The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this could impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this could impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4) The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this may impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this may impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)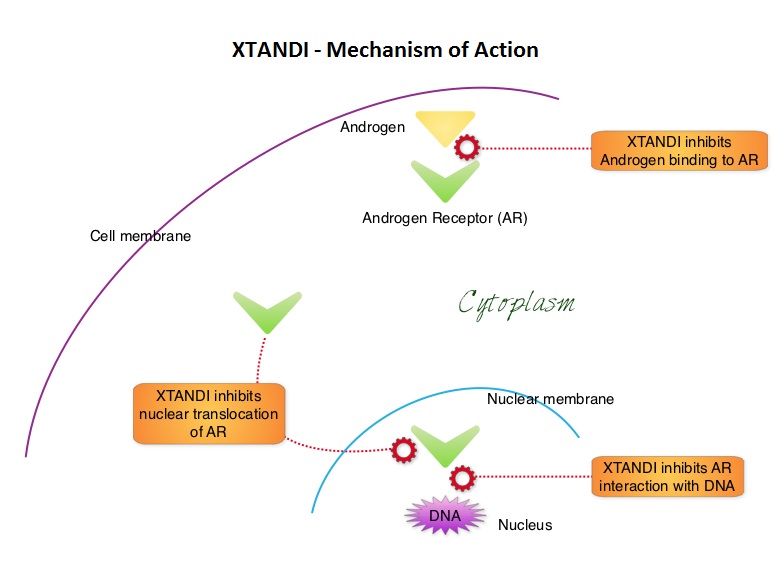 XTANDI® (Enzalutamide) is a second-generation anti-androgen with no reported agonistic effects. It competitively inhibits androgens and AR binding to androgens as well as AR nuclear translocation and interaction with DNA. It thus inhibits several steps in the AR signaling pathway. XTANDI® was first approved by the FDA in 2012, for the treatment of patients with metastatic CRPC who have previously received TAXOTERE® (Docetaxel) based chemotherapy. The PREVAIL study is a double-blind, placebo-controlled, phase III trial in which 1,717 chemotherapy-naive patients with mCRPC (metastatic Castrate Resistant Prostate Cancer) were randomly assigned 1:1 to receive either XTANDI® 160 mg/day or placebo. Prior treatment with surgery or radiation therapy for their primary tumor, as well as hormonal intervention with a LHRH (Luteinizing Hormone Releasing Hormone) agonist or first-generation anti-androgen was allowed. The two co-primary endpoints were Overall Survival (OS) and radiographic Progression Free Survival (rPFS), as measured by bone scans and CT scans. At the time of preplanned interim analysis, XTANDI® demonstrated a statistically significant benefit over placebo with a 30% reduction in risk of death (OS: HR= 0.70; P< 0.0001) and an 81% reduction in risk of radiographic Progression Free Survival (rPFS: HR 0.19; P< 0.0001). Further, the response rates were meaningful with 20% complete responses and 39% partial responses (59% Response Rate) compared with 5% Response Rate in the placebo group (P<0.0001). XTANDI® also significantly delayed the median time to chemotherapy by 17 months compared with those who took placebo (P<0.0001). Based on the results of this interim analysis, the Independent Data Monitoring Committee recommended stopping the study and allowing patients in the placebo group to receive XTANDI®. XTANDI® was well tolerated and the most common side effects were hot flashes, weight gain, fatigue, constipation, back and joint pain. The authors concluded that XTANDI® significantly improves OS and rPFS in patients with chemotherapy-naive mCRPC and can significantly delay the need for chemotherapeutic intervention. Beer TM, Armstrong AJ, Sternberg CN, et al. J Clin Oncol 32, 2014 (suppl 4; abstr LBA1)
XTANDI® (Enzalutamide) is a second-generation anti-androgen with no reported agonistic effects. It competitively inhibits androgens and AR binding to androgens as well as AR nuclear translocation and interaction with DNA. It thus inhibits several steps in the AR signaling pathway. XTANDI® was first approved by the FDA in 2012, for the treatment of patients with metastatic CRPC who have previously received TAXOTERE® (Docetaxel) based chemotherapy. The PREVAIL study is a double-blind, placebo-controlled, phase III trial in which 1,717 chemotherapy-naive patients with mCRPC (metastatic Castrate Resistant Prostate Cancer) were randomly assigned 1:1 to receive either XTANDI® 160 mg/day or placebo. Prior treatment with surgery or radiation therapy for their primary tumor, as well as hormonal intervention with a LHRH (Luteinizing Hormone Releasing Hormone) agonist or first-generation anti-androgen was allowed. The two co-primary endpoints were Overall Survival (OS) and radiographic Progression Free Survival (rPFS), as measured by bone scans and CT scans. At the time of preplanned interim analysis, XTANDI® demonstrated a statistically significant benefit over placebo with a 30% reduction in risk of death (OS: HR= 0.70; P< 0.0001) and an 81% reduction in risk of radiographic Progression Free Survival (rPFS: HR 0.19; P< 0.0001). Further, the response rates were meaningful with 20% complete responses and 39% partial responses (59% Response Rate) compared with 5% Response Rate in the placebo group (P<0.0001). XTANDI® also significantly delayed the median time to chemotherapy by 17 months compared with those who took placebo (P<0.0001). Based on the results of this interim analysis, the Independent Data Monitoring Committee recommended stopping the study and allowing patients in the placebo group to receive XTANDI®. XTANDI® was well tolerated and the most common side effects were hot flashes, weight gain, fatigue, constipation, back and joint pain. The authors concluded that XTANDI® significantly improves OS and rPFS in patients with chemotherapy-naive mCRPC and can significantly delay the need for chemotherapeutic intervention. Beer TM, Armstrong AJ, Sternberg CN, et al. J Clin Oncol 32, 2014 (suppl 4; abstr LBA1)