
The FDA on June 3, 2025, approved NUBEQA® for metastatic Castration-Sensitive Prostate Cancer (mCSPC). The FDA previously approved NUBEQA® in combination with Docetaxel for mCSPC. NUBEQA® is a product of Bayer Healthcare Pharmaceuticals Inc.
Author: RR
Neoadjuvant Niraparib Plus Dostarlimab in BRCA or PALB2-Mutated Triple Negative Breast Cancer: Phase II TBCRC 056 Results
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. It is estimated that in the US, approximately 316,950 new cases of female breast cancer were diagnosed in 2025, and about 42,170 women died of the disease, largely due to metastatic recurrence.
Rationale for a Chemotherapy-Free Neoadjuvant Strategy
Patients with germline BRCA1/2 or PALB2–mutated breast cancer represent a biologically distinct population with heightened sensitivity to PARP inhibition. Beyond synthetic lethality, accumulating preclinical evidence suggests that PARP inhibitors activate the cGAS/STING pathway, increasing intratumoral inflammation, recruiting CD8+ T cells, and potentially priming tumors for immune checkpoint blockade.
While prior studies have not demonstrated a clear benefit for combining PARP inhibitors with immunotherapy in the advanced TNBC (Triple-Negative Breast Cancer) setting, investigators hypothesized that the early-stage, neoadjuvant context, characterized by less immune exhaustion and lower tumor burden, might offer a more permissive environment for synergy.
Study Design and Patient Population
TBCRC 056 is a randomized, Phase II study evaluating the PARP inhibitor Niraparib (ZEJULA®) in combination with the anti–PD-1 antibody Dostarlimab (JEMPERLI®) as neoadjuvant therapy for patients with HER2-negative breast cancer harboring germline BRCA1/2 or PALB2 mutations. The trial includes cohorts for both triple-negative and Estrogen Receptor (ER) positive disease. The current analysis focuses on TNBC cohorts (Arms A and B). Participants with ER positive breast cancer will be placed directly into Arm C. There is no randomization for these participants.
Eligible patients were ≥18 years old with Stage I–III TNBC, primary tumors ≥1.0 cm, HER2-negative disease, and confirmed germline BRCA1/2 or PALB2 mutations. Patients were randomized to:
- Arm A: Niraparib 200 mg orally once daily plus Dostarlimab 500 mg IV every 3 weeks for 18 weeks
- Arm B: Niraparib monotherapy for 3 weeks followed by Niraparib plus Dostarlimab for 15 weeks
Tumor biopsies were obtained at baseline and week 3 to assess immune modulation. Surgery followed 18 weeks of therapy, with optional additional neoadjuvant treatment at investigator discretion if residual disease was detected.
RCB (Residual Cancer Burden in the breast tissue and axillary lymph nodes) Categories:
- RCB 0:No invasive cancer cells found (pCR).
- RCB I (Minimal):Very small amount of residual disease.
- RCB II (Moderate):Moderate amount of residual disease.
- RCB III (Extensive):Significant amount of residual disease.
Endpoints and Statistical Considerations
The Primary endpoints were:
- Pathologic Complete Response (pCR; RCB-0) rate in Arms A and B combined
- Change in stromal Tumor-Infiltrating Lymphocytes (sTILs) from baseline to week 3
The study was powered to detect a pCR rate of ≥50%, allowing rejection of a null hypothesis pCR rate <30%.
Baseline Characteristics
A total of 46 patients with TNBC were enrolled across Arms A and B. The median age was 39.3 years, reflecting the young demographic typical of germline BRCA-associated disease. Most patients were White (84.8%), with representation from Black, Asian, and Hispanic populations. Clinically, 37.0% had Stage I disease, 45.7% Stage II, and 17.4% Stage III. The majority had node-negative and high-grade (grade 3) tumors. BRCA1 mutations predominated (82.6%), with the remainder harboring BRCA2 mutations. No PALB2-mutated TNBC patients were enrolled in this cohort.
Efficacy Outcomes: pCR and Residual Disease
Neoadjuvant Niraparib plus Dostarlimab achieved a pCR rate of 50.0% (90% CI, 37.1%–62.9%) among evaluable patients, meeting and exceeding the study’s predefined efficacy threshold.
Notably:
- pCR rates were identical in both treatment strategies, at 50% in Arm A (concurrent therapy) and Arm B (niraparib lead-in)
- The combined RCB-0/I rate was 60.0%, suggesting meaningful tumor eradication or minimal residual disease in a majority of patients
- Approximately 24% of patients crossed over to additional preoperative therapy, reflecting real-world decision-making when residual disease is identified
These findings support the robustness of the regimen regardless of initial PARP inhibitor lead-in.
Immune Modulation and Biomarker Insights
A key translational objective of TBCRC 056 was to characterize early immune changes within the tumor microenvironment.
Both treatment arms demonstrated statistically significant increases in sTILs from baseline to week 3:
- Arm A: Mean sTILs increased from 16% to 27.4%
- Arm B: Mean sTILs increased from 19.5% to 42.1%, suggesting a pronounced immune activation following PARP inhibitor exposure
Importantly, patients who achieved pCR had higher baseline sTIL levels than those who did not, underscoring the prognostic relevance of preexisting immune infiltration. Baseline sTILs were also associated with achieving RCB-0/I.
In contrast, baseline PD-L1 expression, estrogen receptor status (ER-0 vs ER-low), and short-term changes in sTILs were not independently associated with pCR, highlighting the complexity of immune–genomic interactions in BRCA-driven TNBC.
Safety and Tolerability
The safety profile of Niraparib plus Dostarlimab was consistent with known toxicities of PARP inhibition and immune checkpoint blockade.
- Grade ≥2 treatment-related adverse events occurred in 82.6% of patients
- Grade 3 events were reported in 26.1%, and grade 4 events were rare (2.2%)
- The most common higher-grade toxicities included anemia, fatigue, hypertension, hypothyroidism, and neutropenia
Treatment discontinuation occurred in 13% of patients, with discontinuations split between Niraparib and Dostarlimab, suggesting manageable but clinically relevant toxicity in a neoadjuvant setting.
Key Takeaways for Oncology Practice
- TBCRC 056 demonstrates that a chemotherapy-free neoadjuvant therapy with Niraparib combined with Dostarlimab achieved a 50% pathologic Complete Response (pCR) rate in patients with germline BRCA-mutated early-stage TNBC, exceeding the study’s predefined efficacy threshold.
- pCR rates were identical whether Dostarlimab was administered concurrently with Niraparib or following a short PARP inhibitor lead-in, suggesting flexibility in treatment sequencing.
- Treatment was associated with a significant increase in stromal Tumor-Infiltrating Lymphocytes (sTILs) within 3 weeks, supporting biologic synergy between PARP inhibition and PD-1 blockade in early-stage disease.
- Higher baseline sTIL levels were associated with both pCR and minimal residual disease (RCB-0/I), whereas baseline PD-L1 expression and ER-low status were not predictive.
- These findings support further investigation of biomarker-driven, non-chemotherapy neoadjuvant strategies in genetically defined TNBC populations.
TBCRC-056: A phase II study of neoadjuvant niraparib with dostarlimab for patients with BRCA- or PALB2-mutated breast cancer: results from the TNBC cohorts. Mayer EL, Graham N, Leon-Ferre RA, et al. Presented at: 2025 San Antonio Breast Cancer Symposium; December 9-12, 2025; San Antonio, TX. Abstract RF5-02.
Late Breaking Abstract – ASH 2025: Teclistamab Plus Daratumumab Redefines Outcomes in Early Relapsed Multiple Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 36,110 new cases will be diagnosed in 2025, and 12,030 patients are expected to die of the disease. Multiple Myeloma is a disease of the elderly, with a median age at diagnosis of 69 years and characterized by intrinsic clonal heterogeneity. Almost all patients eventually will relapse, and patients with a high-risk cytogenetic profile, extramedullary disease or refractory disease have the worst outcomes. The introduction of Proteasome Inhibitors, Immunomodulatory agents and CD38 targeted therapies has resulted in higher Response Rates, as well as longer Progression Free Survival (PFS) and Overall Survival (OS), with the median survival for patients with myeloma approaching 10 years or more. Nonetheless, multiple myeloma in 2025 remains an incurable disease.
Relapsed or Refractory Multiple Myeloma (RRMM) remains a complex clinical challenge, even as therapeutic options continue to expand. Progressive immune dysfunction, cumulative treatment toxicity, and repeated relapses often limit the durability of benefit with conventional salvage regimens. Moreover, the increasingly effective frontline landscape has raised the bar for second- and later-line therapy, leaving fewer highly active, well-tolerated options for patients early in relapse.
BCMA-directed therapies have transformed expectations in advanced disease, particularly with CAR-T cell approaches demonstrating deep responses and prolonged disease control. However, manufacturing timelines, resource intensity, and patient fitness requirements limit universal access. Consequently, there is a critical need for off-the-shelf, immunotherapy-based regimens that deliver CAR-T–like efficacy with broader applicability.
Teclistamab (TECVAYLI®), a bispecific T-cell engaging antibody targeting CD3 on T cells and BCMA on myeloma cells, has previously shown meaningful and durable responses in heavily pretreated RRMM. Daratumumab (DARZALEX®), an anti-CD38 monoclonal antibody, remains a foundational therapy across all disease stages, offering both direct antimyeloma activity and immune modulation. Preclinical and clinical observations suggest that Daratumumab-mediated depletion of immunosuppressive cellular subsets enhances T-cell fitness, providing a strong biological rationale for combination with BCMA-directed bispecific antibodies.
The MajesTEC-3 trial was designed to test whether combining Teclistamab with Daratumumab could improve outcomes compared with established Daratumumab-based regimens in patients with earlier-line RRMM.
Study Design and Patient Population
MajesTEC-3 (NCT05083169) is an ongoing, randomized, open-label, Phase 3 trial conducted across 150 centers in 20 countries. Eligible patients had relapsed or refractory multiple myeloma after one to three prior lines of therapy, including prior exposure to both an immunomodulatory agent and a proteasome inhibitor. Patients with prior BCMA-directed therapy or anti-CD38–refractory disease were excluded.
A total of 587 patients were randomized 1:1 to receive either:
- Teclistamab plus subcutaneous Daratumumab, or
- Investigator’s choice of standard Daratumumab-based therapy, consisting of Daratumumab and Dexamethasone combined with either Pomalidomide (DPd) or Bortezomib (DVd).
Randomization was stratified by choice of control regimen, International Staging System stage, prior exposure to anti-CD38 antibodies, and number of prior treatment lines. The median patient age was approximately 64–65 years, with a median of two prior lines of therapy. Importantly, more than one-third of enrolled patients had high-risk cytogenetic features, reflecting a clinically relevant population.
Treatment Administration: A Patient-Centered, Steroid-Sparing Approach
Patients in the investigational arm received subcutaneous Teclistamab using a step-up dosing strategy, followed by a progressively extended dosing interval, transitioning to monthly administration from cycle 7 onward. Daratumumab was administered subcutaneously according to its approved schedule.
Notably, the regimen became steroid-free after cycle 1, an important quality-of-life consideration for patients requiring long-term therapy. Infection prophylaxis, immunoglobulin supplementation, and monitoring of IgG levels were mandated, with protocol amendments reinforcing best practices for infection prevention during BCMA-directed therapy. The Primary end point was Progression-Free Survival (PFS), as assessed by an Independent Review Committee.
Primary Endpoint: Striking Improvement in Progression-Free Survival
At a median follow-up of 34.5 months, Teclistamab plus Daratumumab demonstrated a highly significant and clinically transformative improvement in PFS compared with DPd or DVd.
- The estimated 36-month PFS rate was 83.4% with Teclistamab–Daratumumab versus 29.7% with standard Daratumumab-based therapy.
- This translated into an 83% reduction in the risk of disease progression or death (HR 0.17; 95% CI, 0.12–0.23; P<0.001).
- The prespecified boundary for superiority was crossed at the first interim analysis.
Importantly, the PFS advantage was consistent across all prespecified and clinically relevant subgroups, including patients with high-risk cytogenetics and those treated in earlier versus later relapse.
Depth and Durability of Response
Beyond delaying progression, Teclistamab–Daratumumab induced exceptionally deep and durable responses:
- Complete Response or better was achieved in 81.8% of patients receiving the combination, compared with 32.1% in the control arm.
- Overall Response Rates were also higher (89.0% vs. 75.3%).
- Rates of Minimal Residual Disease negativity at a sensitivity of 10⁻⁵ were more than threefold higher with Teclistamab–Daratumumab (58.4% vs. 17.1%).
Responses occurred rapidly, with a median time to first response of just over one month, and deepened over time. At three years, nearly 90% of responders in the investigational arm remained in response, suggesting the emergence of a plateau in disease control.
Overall Survival and Symptom Outcomes
Although follow-up for overall survival continues, early analyses favored Teclistamab–Daratumumab, with a high proportion of patients remaining alive beyond two years. Improvements were also observed in time to worsening of myeloma-related symptoms, underscoring the regimen’s clinical and patient-reported benefit.
Safety and Tolerability: Manageable With Established Protocols
The safety profile of Teclistamab–Daratumumab was consistent with the known risks of BCMA-directed bispecific antibodies and Daratumumab. Serious adverse events occurred more frequently in the investigational arm, driven primarily by cytopenias and infections.
- Cytokine Release Syndrome was common but predominantly low grade and largely confined to the step-up dosing period.
- Importantly, the incidence of CRS was lower than that reported with Teclistamab monotherapy, supporting a favorable interaction between the two agents.
- Fatal adverse events were infrequent and decreased following protocol-reinforced infection-prevention strategies.
The trial highlights the critical importance of early immunoglobulin replacement, antimicrobial prophylaxis, and vigilant monitoring, now well established in guidelines for patients receiving BCMA-targeted therapies.
Context Within the Evolving Treatment Landscape
The magnitude of benefit observed with Teclistamab–Daratumumab is particularly notable given the strong performance of the control arm, which exceeded historical expectations from prior DPd and DVd studies. Even in this context, the combination delivered superior depth, durability, and consistency of response. As CAR-T therapies move earlier in the disease course, off-the-shelf immunotherapies such as Teclistamab–Daratumumab offer a complementary strategy, one that combines accessibility, scalability, and sustained disease control. Monthly dosing after the initial treatment phase further supports feasibility in community oncology settings.
Clinical Implications
The MajesTEC-3 trial establishes Teclistamab plus Daratumumab as a highly effective immunotherapy-based option for patients with early relapsed multiple myeloma, delivering unprecedented Progression-Free Survival and deep molecular responses without the logistical barriers of cellular therapy. With appropriate supportive care and infection-prevention strategies, this regimen may meaningfully reset expectations for long-term disease control in a population historically characterized by inevitable relapse.
Conclusion
In patients with multiple myeloma who had received one to three prior lines of therapy, Teclistamab combined with Daratumumab significantly outperformed established Daratumumab-based regimens, offering durable disease control, deep responses, and a manageable safety profile. These findings position Teclistamab–Daratumumab as a potential new standard in earlier-line Relapsed or Refractory Multiple Myeloma, and signal continued progress toward prolonged survival in this traditionally incurable disease.
Teclistamab plus Daratumumab in Relapsed or Refractory Multiple Myeloma. Costa LJ, Bahlis NJ, Perrot A, et al. for the MajesTEC-3 Trial Investigators. N Engl J Med. Published December 9, 2025. DOI: 10.1056/NEJMoa2514663
Late Breaking Abstract – ESMO 2025: Advancing First-Line Therapy in High-Risk NMIBC: Final Results from the Phase III POTOMAC Trial
SUMMARY: The American Cancer Society estimates that 84,870 new cases of bladder cancer will be diagnosed in 2025 and 17,420 will die of the disease. Bladder cancer is the fourth most common cancer in men but is less common in women and the average age at the time of diagnosis is 73 years. Caucasians are more likely to be diagnosed with bladder cancer than African Americans or Hispanic Americans.
Persistent Unmet Need in BCG-Naïve High-Risk NMIBC
High-risk Non–Muscle-Invasive Bladder Cancer (NMIBC) remains a clinically challenging disease despite decades of experience with intravesical Bacillus Calmette-Guérin (BCG). Standard management consists of complete TransUrethral Resection of Bladder Tumor (TURBT) followed by BCG induction and maintenance, However, up to 40% of patients experience early recurrence or progression within two years. For those with high-risk recurrence, radical cystectomy is frequently recommended, an intervention associated with substantial morbidity and quality-of-life implications. These limitations have driven interest in immunotherapy-based strategies aimed at improving disease control earlier in the treatment course and potentially delaying or avoiding radical surgery.
Rationale for Combining PD-L1 Blockade with BCG
Durvalumab (IMFINZI®), a monoclonal antibody targeting Programmed Death-Ligand 1 (PD-L1), has demonstrated clinically meaningful benefit in bladder cancer, most notably in the perioperative setting for muscle-invasive disease. Biologic rationale for combining immune checkpoint inhibition with BCG includes immune priming within the bladder microenvironment and the observation that PD-L1 expression may increase with disease progression or BCG resistance. Introducing checkpoint blockade earlier, before immune escape is fully established, may therefore enhance the durability of response to BCG.
POTOMAC Trial Design and Patient Population
POTOMAC (NCT03528694) was a global, randomized, open-label Phase III trial evaluating whether adding Durvalumab to standard BCG therapy improves outcomes in patients with BCG-naïve, high-risk NMIBC. A total of 1,018 patients from more than 120 centers across 12 countries were randomized 1:1:1 following TURBT to one of three treatment arms:
- Durvalumab plus BCG induction and maintenance (N=339)
- Durvalumab plus BCG induction alone (N=339)
- BCG induction and maintenance alone (control– N=340)
Durvalumab was administered at 1,500 mg IV every four weeks for 13 cycles (one year), while intravesical BCG induction therapy was weekly for 6 weeks and maintenance therapy consisted of three doses at weekly intervals at 3, 6, 12, 18, and 24 months. Patients were stratified by Carcinoma in Situ (CIS) and higher-risk papillary disease. The Primary endpoint was investigator-assessed Disease-Free Survival (DFS) comparing Durvalumab plus BCG induction and maintenance versus BCG alone.
Durable Improvement in Disease-Free Survival
At a median follow-up of 60.7 months, POTOMAC met its primary endpoint. The addition of one year of Durvalumab to BCG induction and maintenance resulted in a 32% reduction in the risk of high-risk disease recurrence or death compared with BCG alone (HR=0.68; P=0.015). Disease-free survival curves separated early and remained consistently apart over time, underscoring both early and sustained benefit. The median DFS was not reached in either arm. Importantly, Durvalumab combined with BCG induction alone, without maintenance BCG, did not improve outcomes, reinforcing the central role of adequate BCG exposure in disease control.
Overall Survival and Long-Term Follow-Up
Although the study was not powered to detect Overall Survival differences, extended follow-up showed no evidence of harm associated with Durvalumab. Descriptive analyses suggested numerically favorable survival outcomes with the combination regimen, providing reassurance regarding long-term safety in this curative-intent population.
Safety Profile and Treatment Tolerability
The safety profile of Durvalumab plus BCG was consistent with the known toxicities of each agent. Grade 3 or 4 treatment-related adverse events occurred more frequently with combination therapy than with BCG alone, but these events were generally manageable. No treatment-related deaths were reported. Common adverse effects reflected expected urinary and immune-related events, supporting the feasibility of integrating systemic immunotherapy into NMIBC management.
Context within the Evolving NMIBC Landscape
POTOMAC represents one of the longest follow-up datasets evaluating immune checkpoint inhibition in NMIBC and adds to a growing body of evidence supporting this strategy. Together with prior positive trials exploring PD-1/PD-L1 inhibitors alongside BCG, the data suggest that immune checkpoint blockade can meaningfully augment standard therapy when combined with full-course BCG. Differences among trials highlight the importance of patient selection, adequate maintenance therapy, and sufficient duration of treatment exposure.
Clinical Implications for Practice
The POTOMAC findings reinforce several key principles for clinicians:
- Maintenance BCG remains essential and should not be replaced by systemic immunotherapy alone
- Early integration of immune checkpoint blockade can improve disease control in carefully selected high-risk patients
- Long-term follow-up matters, particularly in NMIBC where durable bladder preservation is a primary goal
Conclusion
For patients with BCG-naïve, high-risk NMIBC, the addition of one year of Durvalumab to standard BCG induction and maintenance delivers a statistically significant and clinically meaningful improvement in Disease-Free Survival with a manageable safety profile. POTOMAC raises the bar for first-line NMIBC therapy and positions combined systemic and intravesical immunotherapy as a compelling new option for this high-risk population.
Durvalumab in combination with BCG for BCG-naive, high-risk, non-muscle-invasive bladder cancer (POTOMAC): final analysis of a randomised, open-label, phase 3 trial. De Santis M, Redorta JP, Nishiyama H, et al. The Lancet. 2025;406:2221-2234.
Reassessing First-Line Chemotherapy Selection in Metastatic PDAC: Insights from the PASS-01 Trial
SUMMARY: The American Cancer Society estimates that in 2025, about 67,440 people will be diagnosed with pancreatic cancer and 51,980 people will die of the disease. Pancreatic Ductal AdenoCarcinoma (PDAC) remains one of the most lethal malignancies, with most cases diagnosed at advanced stages and few modifiable risk factors identified to date.
Pancreatic ductal adenocarcinoma is characterized by marked biological heterogeneity and limited therapeutic durability. While combination chemotherapy regimens have modestly extended survival in the metastatic setting, outcomes remain poor for the majority of patients, underscoring the urgent need for better treatment selection strategies. Molecular stratification has emerged as a promising approach in PDAC, supported by well-established predictive biomarkers such as germline BRCA2 and PALB2 alterations, which identify a subset of patients more likely to benefit from platinum-based chemotherapy and PARP inhibition. Beyond DNA damage repair defects, transcriptomic profiling has further refined the molecular landscape of PDAC, consistently identifying two dominant expression subtypes-Classical and Basal-like, with important prognostic and potentially predictive implications.
The Classical subtype is generally associated with a more differentiated epithelial phenotype and improved survival, whereas Basal-like tumors exhibit stem-like features, relative chemoresistance, and inferior outcomes. Prior nonrandomized and prospective studies have suggested differential chemotherapy sensitivity between these subtypes, raising the question of whether transcriptional classification could inform first-line regimen selection. The Pancreatic Adenocarcinoma Signature Stratification for Treatment-01 (PASS-01) trial was designed to address this gap by prospectively comparing two commonly used first-line chemotherapy regimens, modified FOLFIRINOX (mFOLFIRINOX) and Gemcitabine plus nab-Paclitaxel (GnP), while embedding comprehensive molecular and translational analyses.
PASS-01 Trial Design and Study Objectives
PASS-01 was a randomized, open-label, multinational Phase II study conducted across centers in Canada and the United States. The trial enrolled patients with de novo metastatic PDAC who were chemotherapy-naïve between October 2020 and January 2024, and excluded individuals harboring germline pathogenic variants in BRCA1, BRCA2, or PALB2, thereby removing a population with known platinum sensitivity. Patients were randomized 1:1 to receive either mFOLFIRINOX (N=80) or GnP (N=80. The Primary endpoint was Progression-Free Survival (PFS) in the intention-to-treat population, using a relaxed significance threshold appropriate for a signal-seeking Phase II design. Key Secondary objectives included Overall Survival (OS), Safety, Objective Response Rates (ORR), and exploratory analyses evaluating outcomes according to RNA expression subtype, GATA6 expression, Patient-Derived Organoid (PDO) data, and other molecular correlatives.
Importantly, PASS-01 incorporated mandatory pretreatment tumor biopsies whenever feasible. These samples underwent whole-genome and transcriptome sequencing, RNA-based subtype classification, and PDO generation, with results reviewed in a molecular tumor board to inform later-line treatment decisions. This design allowed for a real-world assessment of the feasibility and clinical relevance of upfront molecular profiling in metastatic PDAC.
First-Line Efficacy Outcomes in the Overall Study Population
With a median follow-up of 8.3 months, PFS was numerically longer with GnP compared with mFOLFIRINOX, although the difference did not reach conventional statistical significance. Median PFS in the intention-to-treat population was 5.3 months with GnP versus 4.0 months with mFOLFIRINOX. Similar trends were observed in the per-protocol analysis.
Overall Survival outcomes favored GnP more clearly. Median OS approached 10 months with GnP and was under 9 months with mFOLFIRINOX, translating into a statistically significant hazard ratio favoring the Gemcitabine-based regimen. Notably, these differences persisted after adjustment for key clinical covariates, including performance status, liver metastases, and KRAS mutation status. While absolute survival gains were modest, these findings are clinically relevant given the lack of head-to-head randomized data comparing these regimens in Western populations. Objective Response Rates were comparable between treatment arms. However, Disease Control Rate and Durability of Response favored GnP. Patients treated with GnP experienced a higher Disease Control Rate and a longer Duration of Response, suggesting more sustained benefit in a subset of patients.
Safety Profile and Treatment Tolerability
Treatment-related toxicity differed meaningfully between regimens. Hospitalizations due to adverse events were more frequent in the mFOLFIRINOX arm, driven primarily by gastrointestinal complications, febrile neutropenia, and serious infections. In contrast, severe toxicities with GnP were less common and more limited in scope. These safety differences are particularly relevant in a population with aggressive disease biology and limited physiologic reserve, where treatment tolerability may influence both quality of life and the ability to receive subsequent therapy.
Impact of Transcriptional Subtypes on Clinical Outcomes
One of the most informative aspects of PASS-01 was its prospective evaluation of RNA expression subtypes. Among patients with adequate tissue for analysis, approximately 75% were classified as Classical and 25% as Basal-like, consistent with prior reports. Across the entire cohort, Basal-like tumors were associated with numerically shorter PFS and OS compared with Classical tumors, reinforcing their adverse prognostic significance.
When outcomes were examined by treatment arm within each subtype, important patterns emerged. In patients with Classical PDAC, PFS was similar between regimens, but OS was notably longer with GnP compared with mFOLFIRINOX. Conversely, in Basal-like disease, outcomes were uniformly poor regardless of regimen, though trends consistently favored GnP across PFS, Response Rate, and Duration of Response. These findings suggest that Basal-like tumors may derive limited benefit from intensified multi-agent chemotherapy and may be particularly resistant to Fluorouracil and Irinotecan-based approaches.
GATA6 Expression as a Pragmatic Surrogate Biomarker
Given prior evidence linking GATA6 expression with the Classical subtype, PASS-01 also evaluated GATA6 RNA in situ hybridization as a pragmatic surrogate biomarker. High GATA6 expression correlated strongly with Classical transcriptional identity. While patients with high GATA6 expression demonstrated a trend toward longer PFS, GATA6 status alone did not reliably predict differential benefit from mFOLFIRINOX versus GnP. These findings suggest that while GATA6 may serve as a useful prognostic marker, its role as a standalone predictive tool for chemotherapy selection remains limited and may require integration into broader multiplex or composite biomarker platforms.
Early CA 19-9 Dynamics as a Biomarker of Treatment Response
PASS-01 also provided important insights into the utility of early CA 19-9 changes as a biomarker of treatment response. Among patients with evaluable markers, a decline in CA 19-9 within four weeks of therapy initiation was associated with significantly prolonged PFS, whereas early increases were linked to inferior outcomes. However, a subset of patients with early CA 19-9 rises subsequently achieved radiographic disease control, underscoring that CA 19-9 kinetics should not be used in isolation to prompt premature treatment discontinuation. These findings support the potential role of early biomarker dynamics, particularly when combined with emerging tools such as circulating tumor DNA, in adaptive treatment strategies.
Translational Findings and the Challenge of Second-Line Therapy
Despite the extensive molecular profiling and use of correlate-guided recommendations, outcomes in the second-line setting were uniformly poor. Only about half of patients were able to receive subsequent therapy, and survival following progression was measured in months. Correlate-guided treatment selection did not meaningfully improve outcomes compared with standard approaches, highlighting the clinical reality that opportunities for precision intervention in PDAC are often lost once patients progress beyond first-line therapy.
Clinical Implications for First-Line Treatment Selection
PASS-01 confirms that outcomes with standard first-line combination chemotherapy for metastatic PDAC remain disappointing, even in carefully selected clinical trial populations. Within this context, the modest but consistent efficacy and safety advantages observed with GnP over mFOLFIRINOX are practice-informing, particularly for patients without known DNA repair defects. More importantly, the trial reinforces the prognostic importance of transcriptional subtypes and supports the concept that molecular features should be assessed early, when they are most likely to influence meaningful treatment decisions.
As novel therapeutic strategies, including KRAS-targeted agents and rational combination approaches, move into earlier lines of therapy, transcriptional subtype may prove critical in guiding regimen selection and trial design. PASS-01 demonstrates that comprehensive upfront molecular profiling is feasible in multicenter settings and provides a framework for future biomarker-driven trials aimed at improving first-line outcomes in this highly lethal disease.
Key Takeaways and Conclusions
In the Phase II PASS-01 trial, Progression-Free Survival was similar between mFOLFIRINOX and Gemcitabine plus nab-Paclitaxel. However, Overall Survival, treatment durability, and safety trends favored the Gemcitabine-based regimen. Molecular analyses confirmed the adverse prognosis associated with Basal-like PDAC and suggested limited benefit from intensified chemotherapy in this subgroup. Collectively, these findings emphasize the critical importance of optimizing first-line treatment strategies and integrating molecular stratification early in the disease course, as opportunities for effective intervention rapidly diminish after progression.
PASS-01: Randomized Phase II Trial of Modified FOLFIRINOX Versus Gemcitabine/Nab-Paclitaxel and Molecular Correlatives for Previously Untreated Metastatic Pancreatic Cancer. Knox JJ, O’Kane G, King D, et al. J Clin Oncol. 2025;43:3355-3368.
Therapeutic Prowess and Potential of Multifunctional Therapeutics: A Review of Bispecific Antibodies
Written by: Jaffer A. Ajani, MD, FASCO
This educational opportunity is sponsored by: Jazz Pharmaceuticals
Concept and Technology
Bispecific antibodies (BsAbs) transcend conventional limitations of therapeutic protein engineering by simultaneously engaging two distinct biological targets. Rooted in molecular cooperation, BsAbs combine two functional antigen-binding fragments (often Fab arms) into a single molecule.1 A considerable novelty over traditional monoclonal antibodies (mAbs), which target a single epitope, BsAbs lead to forced cellular proximity or receptor clustering.1–3 Technological challenges of manufacturing BsAbs for optimal pharmacokinetics (PK), stability, and purity remain. Yet, their dual-targeting allows BsAbs to mediate synergistic effects and intervene in complex, multi-factorial disease pathways—for example, in oncology, where we find multiple redundant receptors, ligands, and evasion mechanisms.1 The following review will review BsAb structures, mechanisms of action, safety profiles, and future directions.
Structural Variants
- Non-IgG-like (Fc-Silent) variants are characterized by a lack of the Fragment crystallizable (Fc) domain, resulting in small molecules that are rapidly cleared by the kidneys, necessitating frequent dosing. Their advantage is high potency and efficient tissue penetration.4 These include:
- Bispecific T-Cell Engagers (BiTEs): Typically constructed as tandem single-chain variable fragments (scFv) that link a tumor-associated antigen (TAA) binder and a CD3 binder via a peptide linker.4 Blinatumomab is a well-known example that achieves potent cellular redirection.
- Dual Affinity Re-targeting Molecules (DARTs): Similar to BiTEs, DARTs incorporate an additional disulfide bridge to improve structural stability.
- Killer Cell Engagers (BiKEs/TriKEs): These target the innate immune system by engaging CD16 on NK cells. Trispecific Killer Engagers (TriKEs) feature a third component, such as an IL-15 crosslinker, to sustain NK cell proliferation and cytotoxicity.4
- IgG-like (Fc-Containing) formats retain the Y-shaped IgG structure, including the Fc domain, conferring prolonged serum half-life via FcRn recycling.4 However, assembling two different heavy chains and two different light chains into a functional heterodimer without forming undesirable mispaired byproducts demands intensive engineering—e.g., CrossMab and/or Knobs-into-Holes (KiH) technologies.4–6
Mechanisms of Action (MOA)
The therapeutic power of BsAbs lies in their ability to execute mechanisms categorized as acting in-trans or in-cis, based on their molecular or cellular target configuration.
- In-Trans Mechanisms: The core in-trans function is creating a physical linkage between two distinct molecular or cellular entities. These include:
- Cellular Bridging (T-Cell Engagers; TCEs): This is the hallmark of oncology BsAbs. By simultaneously binding a TAA and CD3 on T cells, the BsAb forces a physical link, forming a cytolytic synapse.6 This mechanism bypasses the need for natural T-cell receptor (TCR) clustering and Major Histocompatibility Complex (MHC) presentation, allowing the T cell to attack regardless of the tumor’s MHC status.4,6
- Co-factor Mimicry: Outside of cytotoxicity, BsAbs can direct components to form a functional complex. Emicizumab, approved for Hemophilia A, is an example.2–6
- In-Cis Mechanisms: Involve targeting components that reside on the same cell or act within the same signaling pathway. These include:
- Dual Signaling Inhibition (Dual Blockade): Simultaneously blocks two different receptors or ligands to suppress synergistic pathways crucial for disease progression.
- Examples: Targeting HER2/HER3 (Zenocutuzumab) or EGFR/MET (Amivantamab) to halt parallel proliferation cascades in cancer.1–6
- Biparatopic Engagement: By binding two distinct, non-overlapping epitopes on the same antigen4,5, biparatopics intensify control over one oncogenic “addiction” pathway via geometry-driven clustering, internalization, and boosted Fc effector functions. Biparatopics enhance binding avidity and promote superior functional modulation of the target, such as forced receptor clustering and internalization, the latter being highly beneficial for Antibody-Drug Conjugates (ADCs). Biparatopic binding drives dense clustering of the same receptor, leading to “caps” on the cell surface, resulting in potent receptor internalization and degradation. This yields deeper and more durable signal blockade than a single monoclonal antibody or cocktail.7 The high local receptor and antibody density also enables multimodal effector functions, and helps overcome resistance within a single pathway by engaging distinct functional domains to block both ligand-dependent and ligand-independent signaling and interfere with heterodimerization (e.g., HER2/HER3). They also retain efficacy when tumors escape mono-epitope antibodies through epitope masking or mutation.
- Example: Zanidatamab, which targets two distinct HER2 epitopes, and is unique in its ability to induce receptor clustering and “capping.”8-9
- Dual Signaling Inhibition (Dual Blockade): Simultaneously blocks two different receptors or ligands to suppress synergistic pathways crucial for disease progression.
Clinical Landscape
The BsAb landscape has rapidly expanded since the first approval of Blinatumomab in 2014, reflecting a growing therapeutic impact across multiple disease areas. As of late 2025, fifteen bispecific molecules have secured FDA approval, spanning both oncology and non-oncology indications. This surge underscores the versatility of BsAbs and their ability to address complex biological pathways through innovative mechanisms of action.
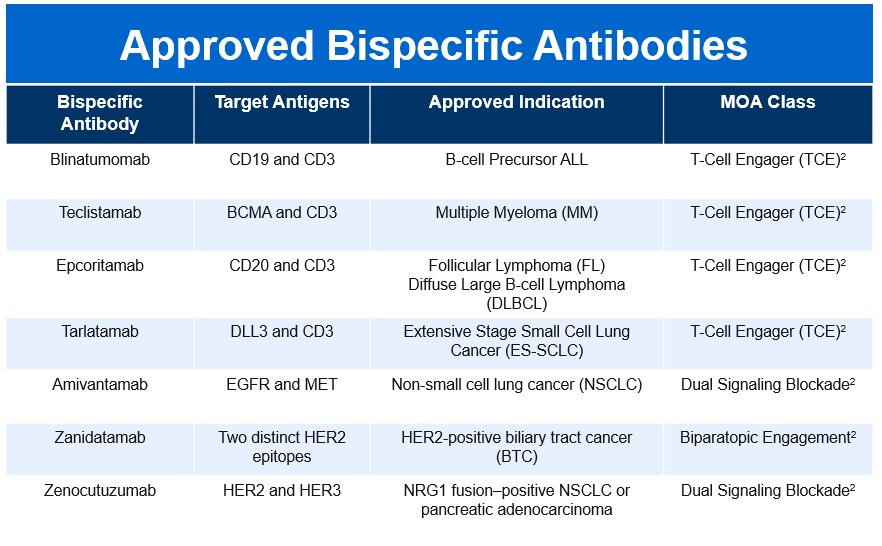
Limitations, Safety, and Risk Mitigation
- Manufacturing Challenges: The inherent complexity of BsAbs introduces challenges related to stability, manufacturability, and impurity control.1 The fusion of exogenous antigen-binding domains can decrease biophysical stability, and the complex assembly process frequently results in the formation of product-related impurities and mispaired species, which are difficult to remove during purification. These factors are not merely manufacturing hurdles; they directly influence the biological activity and, critically, the immunogenic potential of the final drug product.
- MOA-Specific Toxicity: The safety profile of BsAbs is highly dependent on their MOA
- T-Cell Engager Toxicity: The highly potent, acute T-cell activation triggered by TCEs results in two major, distinct safety concerns. The first is Cytokine Release Syndrome (CRS), a serious acute toxicity caused by the mass release of systemic cytokines. CRS has been reported in up to 70% of patients receiving BsAbs, often necessitating hospitalization and precise management protocols. Severe cases (Grade ≥ 3) occur in 5–10% of patients.6 The second concern is Neurotoxicity (ICANS), which, while less frequent than CRS, affects 10–15% of patients and can range from mild confusion to cerebral edema.6 In addition, there is an Immune Regulation Paradox. Paradoxically, T-BsAb therapy can trigger the expansion and activation of inhibitory Regulatory T (Treg) cells in the tumor microenvironment, leading to the production of anti-inflammatory cytokines like IL-10. This critically inhibits the desired effector T-cell response, suggesting that combination strategies—such as transient Treg ablation—may be necessary to maximize efficacy.6
- Pathway Blocker and Biparatopic Toxicity: These agents generally do not induce acute, systemic cytokine surges. Instead, their adverse event profiles reflect the targeted receptors. For example, the dual signaling blocker Amivantamab (targets EGFR/MET) exhibits EGFR-inhibition-related dermatologic toxicities, like paronychia, skin fissures, and pruritus, as well as infusion reactions. Dual checkpoint inhibitors can have classic IO toxicities. Biparatopic antibodies, like Zanidatamab, demonstrate a manageable profile but frequently cause gastrointestinal toxicities (such as diarrhea and nausea/vomiting) and infusion-related reactions (IRRs).9 Importantly, clinical data for Zanidatamab confirmed no reports of CRS.9
- Mitigating Immunogenicity Risk: The complex structures, engineered sequences, and immunostimulatory MOAs of oncology BsAbs contribute to an increased risk of immunogenicity compared to mAbs. Mitigation must begin at the engineering stage, utilizing in silico prediction and in vitro assays to guide the selection of low-risk antibody constructs through deimmunization and tolerization methods.
Future Directions
The BsAb pipeline remains robust, reflecting a continuous drive toward addressing current clinical limitations and expanding into novel biological territories. The future of BsAbs is characterized by a strategic shift toward overcoming the immunosuppressive tumor microenvironment (TME). Emerging candidates are now focused on targets that modulate the innate immune system and TME suppression, such as LILRB1/2 bispecific IgG1 antibodies for advanced solid tumors.10-11 Furthermore, BsAbs are expanding beyond simple blockade, with molecules like SAR446422 (CD28xOX40 bispecific) in trial for inflammatory indications, demonstrating the potential for BsAbs to achieve synergistic co-stimulatory agonism.10-11 The continuous innovation in structural design, focused now on minimizing impurity-driven immunogenicity and maximizing the therapeutic window, ensures that BsAbs are poised to become the standard for highly tailored, multifunctional therapeutic intervention across diverse and complex diseases. The future of BsAbs is very promising.10-11
References:
- Shan KS, Musleh Ud Din S, Dalal S, Gonzalez T, Dalal M, Ferraro P, Hussein A, Vulfovich M. Bispecific Antibodies in Solid Tumors: Advances and Challenges. International Journal of Molecular Sciences. 2025; 26(12):5838. https://doi.org/10.3390/ijms26125838.
- The Bispecific 2024 Landscape Review. Beacon Intelligence. 2024. https://beacon-intelligence.com/landscape-reviews/bispecific/. Accessed November 23, 2025.
- Ai Z, Wang B, Song Y, Cheng P, Liu X, Sun P. Prodrug-based bispecific antibodies for cancer therapy: advances and future directions. Front Immunol. 2025;16:1523693. Published 2025 Jan 22. doi:10.3389/fimmu.2025.1523693.
- Amash A, Volkers G, Farber P, et al. Developability considerations for bispecific and multispecific antibodies. MAbs. 2024;16(1):2394229. doi:10.1080/19420862.2024.2394229.
- Shui L, Wu D, Yang K, Sun C, Li Q, Yin R. Bispecific antibodies: unleashing a new era in oncology treatment. Mol Cancer. 2025;24(1):212. Published 2025 Aug 4. doi:10.1186/s12943-025-02390-y.
- Dewaele L, Fernandes RA. Bispecific T-cell engagers for the recruitment of T cells in solid tumors: a literature review. Immunother Adv. 2025;5(1):ltae005. Published 2025 Jan 27. doi:10.1093/immadv/ltae005.
- Kast F, Schwill M, Stüber JC, et al. Engineering an anti-HER2 biparatopic antibody with a multimodal mechanism of action. Nat Commun. 2021;12(1):3790. Published 2021 Jun 18. doi:10.1038/s41467-021-23948-6.
- Elimova E, Ajani J, Burris H, et al. Zanidatamab plus chemotherapy as first-line treatment for patients with HER2-positive advanced gastro-oesophageal adenocarcinoma: primary results of a multicentre, single-arm, phase 2 study. Lancet Oncol. 2025;26(7):847-859. doi:10.1016/S1470-2045(25)00287-6.
- Ziihera Safety Information. Ziihera HCP (Jazz Pharmaceuticals). https://www.ziiherahcp.com/safety. Accessed November 23, 2025.
- Wen J, Cui W, Yin X, et al. Application and future prospects of bispecific antibodies in the treatment of non-small cell lung cancer. Cancer Biol Med. 2025;22(4):348-375. doi:10.20892/j.issn.2095-3941.2024.0470.
- Engineering the Next Generation of Bispecific Antibodies. PEGS Europe 2024 Archive. https://www.pegsummiteurope.com/24/engineering-bispecifics. Accessed November 23, 2025
FDA Approves Pirtobrutinib in Relapsed/Refractory CLL/SLL
SUMMARY: The FDA on December 3, 2025 granted traditional approval to Pirtobrutinib (JAYPIRCA®) for adults with relapsed or refractory Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma (CLL/SLL) who have previously been treated with a covalent BTK inhibitor. In 2023, FDA granted accelerated approval to Pirtobrutinib for adults with CLL/SLL who have received at least two prior lines of therapy, including a BTK inhibitor and a BCL-2 inhibitor.
The American Cancer Society estimates that for 2025, about 23,690 new cases of Chronic Lymphocytic Leukemia (CLL) will be diagnosed in the US and 4460 patients will die of the disease. CLL accounts for about one-quarter of the new cases of leukemia. The average age of patients diagnosed with CLL is around 70 years, and is rarely seen in people under age 40, and is extremely rare in children. Patients with CLL often receive continuous therapy with either Brutons Tyrosine Kinase (BTK) inhibitor, time limited therapy with BCL2 inhibitor Venetoclax given along with anti-CD20 antibody Obinutuzumab, or under certain circumstances, chemoimmunotherapy.
Brutons Tyrosine Kinase (BTK) is a member of the Tec family of kinases, downstream of the B-cell receptor, and is predominantly expressed in B-cells. It is a mediator of B-cell receptor signaling in normal and transformed B-cells. BTK inhibitors inhibit cell proliferation and promote programmed cell death (Apoptosis) by blocking B-cell activation and signaling. BTK is a validated molecular target found across numerous B-cell leukemias and lymphomas including Chronic Lymphocytic Leukemia (CLL), Mantle Cell Lymphoma (MCL), and Waldenstrom Macroglobulinemia (WM).
The 3 covalent BTK inhibitors (cBTKi) presently approved by the FDA for CLL/SLL include Ibrutinib (IMBRUVICA®) Acalabrutinib (CALQUENCE®), and Zanubrutinib (BRUKINSA®). Although covalent BTK inhibitors have dramatically improved outcomes for patients with CLL or SLL, they are not curative. Despite the efficacy of covalent BTK inhibitors, treatment failure often occurs through development of resistance or intolerance.
Pirtobrutinib (JAYPIRCA®) is a next-generation, highly selective, reversible, non-covalent BTK inhibitor (BTKi), developed to reversibly bind BTK, deliver consistently high target coverage regardless of BTK turnover rate, and preserve activity in the presence of the C481 acquired resistance mutations. Pirtobrutinib is 300 times more selective in BTK inhibition versus 98% of other kinases tested in preclinical studies, and inhibits both wild type and C481-mutant BTK with equal low nM potency, and has favorable oral pharmacology. Pirtobrutinib is well tolerated and demonstrated encouraging efficacy and safety in early-phase studies, leading to FDA accelerated approval in December 2023 for patients with CLL/SLL who have received ≥2 prior lines of therapy, including both a BTKi and a BCL-2 inhibitor.
Relapsed/Refractory (R/R) Chronic Lymphocytic Leukemia (CLL) and Small Lymphocytic Lymphoma (SLL) in the post–covalent Bruton Tyrosine Kinase Inhibitor (cBTKi) setting remains a major therapeutic challenge. No prospective, randomized trials have previously evaluated treatment options in this population, and real-world data suggest poor outcomes, particularly after sequential covalent BTKi and BCL-2 inhibitor exposure. The present FDA approval was based on the BRUIN CLL-321 study.
Study Design
BRUIN CLL-321 is the first global, randomized, multicenter, Phase III trial conducted exclusively in patients with R/R CLL/SLL previously treated with a cBTKi.
- Design: Open-label, 1:1 randomization to Pirtobrutinib 200 mg PO daily (N=119) vs. investigator’s choice (IC) of Idelalisib plus Rituximab (IdelaR-N=82) or Bendamustine plus Rituximab (BR-N=37).
- Population: 238 patients; median 3 prior therapies; 50% had prior Venetoclax; high prevalence of high-risk genomic features (del[17p]/TP53 mutation ~54%, complex karyotype up to 72%).
- Endpoints: Primary endpoint was Independent Review Committee (IRC)–assessed Progression-Free Survival (PFS). Secondary endpoints included Time to Next Treatment or death (TTNT), Overall Survival (OS), Overall Response Rate (ORR), and Safety.
Patients could cross over from IC to Pirtobrutinib upon confirmed progression, and treatment beyond IRC-defined progression was permitted if clinical benefit was maintained.
Efficacy Outcomes
- PFS: Median 14.0 months with Pirtobrutinib vs. 8.7 months with IdelaR/BR (HR = 0.54; P =0.0002), representing a 46% reduction in the risk of progression or death.
- TTNT: Median 24.0 months with Pirtobrutinib vs. 10.9 months with IC (HR = 0.37), reflecting sustained clinical benefit beyond protocol-defined progression in many patients.
- OS: No statistically significant difference at final analysis (HR = 1.09), likely influenced by crossover (75.8% of eligible IC patients switched to Pirtobrutinib).
- Subgroup Benefit: PFS improvement was consistent across key high-risk subgroups, including those with del(17p)/TP53 mutation, complex karyotype, and unmutated IGHV.
Safety Profile
Pirtobrutinib was generally well tolerated:
- Grade ≥3 adverse events (AEs): 57.7% with Pirtobrutinib vs. 73.4% with IC.
- Discontinuations due to AEs: 17.2% vs. 34.9%, respectively.
- Class-related BTKi toxicities (atrial fibrillation, hypertension, major bleeding) were infrequent, with rates comparable to or lower than background incidence in CLL populations.
- No cases of Richter transformation were reported in the Pirtobrutinib arm, versus three in the IC group.
Clinical Implications
The BRUIN CLL-321 trial establishes Pirtobrutinib as a new standard of care option for patients with CLL/SLL previously treated with cBTKi, offering:
- Significant PFS improvement in a population with historically poor prognosis.
- Prolonged TTNT, which may be more reflective of real-world benefit than PFS alone.
- Favorable safety profile supporting long-term tolerability.
These findings also raise important considerations for sequencing strategies, including potential use of Pirtobrutinib prior to Venetoclax in certain patients, pending further prospective data (e.g., BRUIN-322 trial).
Takeaway for Practice:
For CLL/SLL patients progressing after cBTKi therapy, Pirtobrutinib offers a meaningful advancement, providing durable disease control with a manageable toxicity profile. BRUIN CLL-321 delivers the first randomized evidence supporting treatment in this setting, addressing a long-standing gap in the therapeutic landscape.
Phase III Trial of Pirtobrutinib Versus Idelalisib/Rituximab or Bendamustine/Rituximab in Covalent Bruton Tyrosine Kinase Inhibitor–Pretreated Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (BRUIN CLL-321). Sharman JP, Munir T, Grosicki S, et al. J Clin Oncol. 2025;43:2538-2549
FDA Approves Niraparib with Abiraterone Acetate plus Prednisone for BRCA2-Mutated mCSPC
SUMMARY: The FDA on December 12, 2025, approved Niraparib and Abiraterone acetate (AKEEGA®) with prednisone for adults with deleterious or suspected deleterious BRCA2-mutated (BRCA2m) metastatic Castration-Sensitive Prostate Cancer (mCSPC), as determined by an FDA-approved test.
Prostate cancer is the most common cancer in American men with the exclusion of skin cancer, and 1 in 8 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 313,780 new cases of prostate cancer will be diagnosed in 2025 and 35,770 men will die of the disease.
Metastatic Castration-Sensitive Prostate Cancer (mCSPC) is a heterogeneous disease. Despite therapeutic advances, outcomes vary significantly based on underlying tumor biology. Approximately 25% of patients with mCSPC harbor Homologous Recombination Repair (HRR) gene mutations, including BRCA1, BRCA2, CHEK2, CDK12, PALB2, and others. Among these, BRCA1/2 mutations account for nearly half of HRR alterations and are particularly associated with aggressive disease biology, resistance to Androgen Receptor Pathway Inhibitors (ARPIs), and shortened Progression-Free and Overall Survival. The integration of AR-pathway inhibitors such as Abiraterone Acetate plus Prednisone into first-line treatment has meaningfully improved outcomes in the general mCSPC population. However, patients with HRR mutations, especially those with BRCA1/2, derive significantly less benefit from these agents alone, highlighting a substantial unmet clinical need.
Rationale for PARP Inhibition in HRR-Altered Prostate Cancer
Cancer cells with HRR deficiencies are vulnerable to PARP (Poly ADP-Ribose Polymerase) inhibition, which blocks DNA repair pathways and induces synthetic lethality. Prior landmark trials, MAGNITUDE (Niraparib with Abiraterone Acetate plus Prednisone) and TALAPRO-2 (Talazoparib plus Enzalutamide), demonstrated the value of combining PARP inhibitors with ARPIs in Castration-Resistant Prostate Cancer (mCRPC) with HRR mutations. However, whether such a combination could offer meaningful benefit earlier in the disease course, in the castration-sensitive setting, remained unknown, until now.
AMPLITUDE Trial Design and Methods
Study Overview
The AMPLITUDE trial (NCT04497844) is a global, Phase 3, randomized, double-blind, placebo-controlled trial designed to evaluate whether combining the PARP inhibitor Niraparib with Abiraterone Acetate plus Prednisone improves clinical outcomes in patients with mCSPC (metastatic Castration-Sensitive Prostate Cancer) and HRR gene alterations.
Patient Population
- Total enrolled: 696 men with mCSPC and at least one HRR gene mutation (germline or somatic)
- Mutation profile: BRCA1, BRCA2, BRIP1, CDK12, CHEK2, FANCA, PALB2, RAD51B, RAD54L
- BRCA1/2 prevalence: 55.6% of enrolled patients
- Metastatic disease burden: 78% were high-volume M1disease, 87% had de novo M1disease and 16% had prior therapy with Docetaxel.
- Prior therapies allowed:
- 6 months or less of Androgen Deprivation Therapy (ADT)
- 6 cycles or less of Docetaxel
- 45 days or less of prior Abiraterone and Prednisone
Randomization and Treatment Arms
Patients were randomized 1:1 to:
- Experimental arm: Niraparib 200 mg once daily plus Abiraterone acetate 1000 mg daily and Prednisone 5 mg daily (N=348)
- Control arm: Placebo plus Abiraterone acetate 1000 mg along with Prednisone 5 mg daily (N=348)
All patients continued on ADT.
Endpoints
- Primary: Radiographic Progression-Free Survival (rPFS), assessed by investigator
- Secondary: Time to Symptomatic Progression (TSP), Overall Survival (OS), Safety/tolerability
Key Results and Interpretation
Efficacy Outcomes
Radiographic Progression-Free Survival (Primary Endpoint)
- Median rPFS:
- Niraparib plus Abiraterone and Prednisone: Not reached
- Abiraterone and Prednisone alone: 5 months (95% CI, 25.8–NR)
- Hazard ratio: 0.63 (P=0.0001)
- BRCA1/2 subgroup: HR =0.52 (P<0.0001)
This translates into a 37% relative risk reduction in progression or death in the overall population, and a 48% reduction in the BRCA1/2 subgroup, indicating a clear therapeutic effect in genetically defined populations.
Time to Symptomatic Progression
- HR (overall): 0.50 (P<0.0001)
- BRCA1/2 subgroup: HR 0.44 (P=0.0001)
This is clinically meaningful, and delaying symptom onset can preserve quality of life and extend time before additional therapies are needed.
Overall Survival (Interim Analysis)
- HR (overall): 0.79 (95% CI, 0.59–1.04; P=0.10)
- BRCA1/2 subgroup: HR 0.75 (95% CI, 0.51–1.11; P=0.15)
In an exploratory analysis of 323 patients with BRCA2 mutations, the rPFS Hazard Ratio (HR) was 0.46 (95% CI: 0.32, 0.66) with median rPFS not estimable for Niraparib and Abiraterone acetate with prednisone compared with 26 months (95% CI: 18, 28) for placebo and Abiraterone acetate with prednisone.
In an exploratory analysis in 373 patients with non-BRCA2 mutations, the HR for rPFS was 0.88 (95% CI: 0.63, 1.24), indicating that the overall improvement was primarily attributed to the results seen in patients with BRCA2 mutations.
Although OS data are not yet mature, the trend suggests a potential survival benefit with longer follow-up.
Safety Profile
The safety of Niraparib plus Abiraterone and Prednisone was consistent with known profiles of both agents. Grade 3-4 AEs in the Niraparib plus Abiraterone and Prednisone was 75.2% versus 58.9% with Abiraterone and Prednisone alone, with the most common higher Grade 3-4 AEs noted in the Niraparib plus Abiraterone and Prednisone group (Anemia: 29.1% vs 4.6% and Hypertension: 26.5% vs 18.4%). The discontinuation rates due to AEs in the Niraparib plus Abiraterone and Prednisone group was 11.0% vs 6.9% in the Abiraterone and Prednisone group. These AEs were manageable with appropriate monitoring. No new safety signals were identified.
Conclusion
The AMPLITUDE trial marks a milestone and provides robust evidence to support Niraparib plus Abiraterone and Prednisone as a new first-line option in mCSPC patients with BRCA1/2 or other HRR gene mutations. By demonstrating that Niraparib plus Abiraterone and Prednisone improves Progression-Free outcomes in HRR-altered mCSPC, especially those with BRCA mutations, it paves the way for a more personalized, biology-driven approach to therapy in this setting. Ongoing follow-up will determine whether this translates into improved survival, but the current data already support Niraparib plus Abiraterone and Prednisone as a new treatment benchmark for this high-risk subgroup.
Phase 3 AMPLITUDE trial: Niraparib (NIRA) and abiraterone acetate plus prednisone (AAP) for metastatic castration-sensitive prostate cancer (mCSPC) patients (pts) with alterations in homologous recombination repair (HRR) genes. Attard G, Agarwal N, Graff J, et al. J Clin Oncol 43, 2025 (suppl 17; abstr LBA5006)
HER2CLIMB-05: Redefining First-Line Maintenance Therapy in HER2-Positive Metastatic Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. It is estimated that in the US, approximately 316,950 new cases of female breast cancer will be diagnosed in 2025, and about 42,170 women will die of the disease, largely due to metastatic recurrence.
Approximately 15-20% of invasive breast cancers overexpress HER2/neu oncogene, which is a negative predictor of outcomes without systemic therapy. Patients with HER2-positive metastatic breast cancer are often treated with anti-HER2 targeted therapy along with chemotherapy, irrespective of hormone receptor status, and this has resulted in significantly improved treatment outcomes. With advances in systemic therapies for this patient population, the incidence of brain metastases as a sanctuary site has increased. Approximately 50% of patients with HER2-positive metastatic breast cancer develop brain metastases. However, systemic HER2-targeted agents, including Tyrosine Kinase Inhibitors, as well as chemotherapy have limited antitumor activity in the brain. Local therapeutic interventions for brain metastases include neurosurgical resection and Stereotactic or Whole-Brain Radiation Therapy. There is a high unmet need for systemic treatment options to treat established brain metastases and reduce the risk for progression in the Central Nervous System (CNS).
Expanding Options Beyond Standard Maintenance
Despite major advances in the management of Human Epidermal growth factor Receptor 2–positive (HER2+) metastatic breast cancer (MBC), disease progression during maintenance therapy remains a persistent challenge. The long-standing first-line (1L) standard of care, induction with Trastuzumab, Pertuzumab, and a Taxane followed by Trastuzumab plus Pertuzumab maintenance, has delivered durable benefit, yet most patients ultimately relapse within two years. This unmet need is particularly relevant in a modern population increasingly exposed to HER2-targeted therapy in the early-stage setting and enriched for de novo metastatic disease.
HER2CLIMB-05 was designed to test whether intensifying HER2 blockade during the maintenance phase, by adding the highly selective oral HER2 tyrosine kinase inhibitor (TKI) Tucatinib (TUKYSA®), could further delay disease progression while preserving tolerability and quality of life.
Study Design and Patient Population
HER2CLIMB-05 (NCT05132582) is a randomized, double-blind, placebo-controlled, International Phase 3 trial enrolling patients with centrally confirmed HER2+ unresectable locally advanced or metastatic breast cancer. Eligible patients had no evidence of disease progression following 4 to 8 cycles of standard 1L induction therapy with Trastuzumab, Pertuzumab, and a taxane, an ECOG performance status of 0 or 1, and no or asymptomatic brain metastases.
A total of 654 patients were randomized 1:1 to receive Tucatinib (300 mg PO twice daily) or placebo, in combination with Trastuzumab and Pertuzumab administered IV every 21 days. Randomization was stratified by de novo versus recurrent disease, Hormone Receptor (HR) status, and presence or history of brain metastases. Endocrine therapy was permitted for patients with HR-positive disease. The Primary endpoint was investigator-assessed Progression-Free Survival (PFS) per RECIST v1.1. Key Secondary endpoints included Overall Survival (OS), PFS by Blinded Independent Central Review (BICR), Central Nervous System (CNS) PFS, safety, and Patient-Reported Outcomes.
The enrolled population reflects current real-world patterns of HER2+ MBC. All patients were female, with a median age of 54 years. Nearly 70% presented with de novo metastatic disease, over half had HR-positive tumors, and 12.4% had a presence or history of brain metastases at baseline. Most patients had excellent performance status, with nearly two-thirds classified as ECOG 0.
Primary Endpoint Met: Significant and Clinically Meaningful PFS Improvement
At a median follow-up of approximately 23 months, HER2CLIMB-05 met its Primary endpoint. The addition of Tucatinib to Trastuzumab and Pertuzumab resulted in a statistically significant and clinically meaningful improvement in PFS compared with standard maintenance therapy alone. Median PFS was 24.9 months in the Tucatinib arm versus 16.3 months in the control arm, corresponding to a 36% reduction in the risk of disease progression or death (Hazard Ratio [HR], 0.64; P < 0.0001). Results from BICR were consistent, reinforcing the robustness of the primary analysis.
Importantly, the PFS benefit was observed across all prespecified subgroups, including patients with and without brain metastases and those with HR-positive or HR-negative disease. This consistency highlights the broad applicability of Tucatinib-based maintenance therapy in HER2+ MBC.
Early Signals in Overall Survival and CNS Outcomes
Overall Survival data remain immature, with approximately 20% of the required events observed at the time of this primary analysis. Median OS has not yet been reached in either arm, with no evidence of detriment associated with Tucatinib and a favorable trend observed.
While CNS-PFS was not reached in the overall population, patients with baseline brain metastases experienced a numerical improvement with Tucatinib, with median CNS-PFS nearly doubling compared with control (8.5 vs 4.3 months). Although preliminary, these findings align with prior HER2CLIMB data supporting Tucatinib’s activity in CNS disease.
Safety Profile: Consistent and Manageable
The safety profile of Tucatinib in combination with Trastuzumab and Pertuzumab was consistent with known toxicities of HER2-directed regimens. Diarrhea, nausea, and transaminase elevations were the most common treatment-emergent adverse events in the Tucatinib arm, the majority of which were low grade and manageable with supportive care and dose modifications.
Grade ≥3 adverse events were more frequent with Tucatinib, particularly elevated ALT and AST; however, hepatic events were generally asymptomatic, reversible, and occurred early in treatment. Discontinuation of Tucatinib due to adverse events occurred in 13.5% of patients, underscoring the importance of proactive monitoring and toxicity management in clinical practice. No new safety signals were identified.
Positioning HER2CLIMB-05 in an Evolving Treatment Landscape
HER2CLIMB-05 adds to a growing body of evidence supporting maintenance intensification strategies in HER2+ MBC. Alongside recent Phase 3 trials such as PATINA and DESTINY-Breast09, these data emphasize that meaningful improvements in disease control can be achieved beyond traditional chemotherapy-based induction regimens.
Unlike antibody–drug conjugate based approaches, Tucatinib-based maintenance offers a chemotherapy-free option that targets HER2 both extracellularly and intracellularly, with particular relevance for patients with brain metastases or those who may not be ideal candidates for prolonged cytotoxic therapy.
Clinical Implications
The HER2CLIMB-05 primary analysis demonstrates that adding Tucatinib to Trastuzumab and Pertuzumab as 1L maintenance therapy significantly prolongs PFS, extending disease control to more than two years in patients with HER2+ metastatic breast cancer. The benefit was consistent across key subgroups, including HR status and CNS involvement, and was achieved with a manageable and familiar safety profile.
As the HER2+ metastatic treatment paradigm continues to evolve, Tucatinib-based maintenance represents an important new option that may delay progression and postpone the need for subsequent cytotoxic therapy. Ongoing follow-up will clarify the impact on Overall Survival, CNS outcomes, and patient-reported Quality of Life, further informing individualized treatment decisions.
HER2CLIMB-05: A Phase 3 Study of Tucatinib Versus Placebo in Combination with Trastuzumab and Pertuzumab as First-line Maintenance Therapy for HER2+ Metastatic Breast Cancer. Dieras V, Curigliano G, Martin M, et al. on behalf of the HER2CLIMB-05 investigators. J Clin Oncol. DOI: 10.1200/JCO-25-02600
PHAROS: Long-Term Efficacy and Survival Outcomes with Encorafenib Plus Binimetinib in BRAF V600E–Mutant Metastatic NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 21% of all cancer deaths. The American Cancer Society estimates that for 2025, about 226,650 new cases of lung cancer will be diagnosed and 124,730 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers and Adenocarcinoma is now the most frequent histologic subtype of lung cancer.
BRAF V600E mutations occur in approximately 1% to 2% of patients with NSCLC and define a biologically distinct subset for which targeted therapy has become a cornerstone of treatment. Dual inhibition of the MAPK pathway with BRAF and MEK inhibitors is currently recommended by clinical guidelines as the preferred first-line approach for patients with BRAF V600E–mutant metastatic NSCLC (mNSCLC), with immunotherapy and chemotherapy-based regimens serving as alternative options.
The Phase II PHAROS study (NCT03915951) is an ongoing, open-label, single-arm, multicenter trial designed to evaluate the efficacy and safety of Encorafenib (BRAFTOVI®) in combination with Binimetinib (MEKTOVI®) in patients with BRAF V600E–mutant mNSCLC. Eligible patients included both treatment-naïve individuals and those with prior systemic therapy for metastatic disease. All patients received oral Encorafenib 450 mg once daily plus oral Binimetinib 45 mg twice daily, administered in continuous 28-day cycles until disease progression, unacceptable toxicity, or treatment discontinuation.
The Primary endpoint of PHAROS was Objective Response Rate (ORR) as assessed by Independent Radiology Review (IRR), with separate analyses prespecified for treatment-naïve and previously treated cohorts. Key Secondary endpoints included Duration of Response (DOR), Progression-Free Survival (PFS), Overall Survival (OS), Disease Control Rate, Safety, and Tolerability. Exploratory analyses evaluated efficacy across clinically relevant subgroups, including smoking history.
A total of 98 patients were enrolled and treated, including 59 treatment-naïve and 39 previously treated patients. At the March 14, 2025 data cutoff, a small proportion of patients in both cohorts remained on active treatment, reflecting durable disease control in a subset of patients. Median treatment duration was substantially longer in the frontline cohort compared with previously treated patients, with more than 40% of treatment-naïve patients receiving therapy for longer than two years.
PHAROS met its Primary endpoint in both cohorts.
In treatment-naïve patients, the confirmed ORR by IRR was 75%, with responses demonstrating marked durability. Median Duration of Response was 40.0 months, and median PFS reached 30.4 months. After a median follow-up of more than four years for overall survival, median OS was 47.6 months, with an estimated 4-year OS rate of 49%, underscoring the potential for prolonged survival with frontline targeted therapy.
In the previously treated cohort, Encorafenib plus Binimetinib also demonstrated clinically meaningful activity. The ORR was 49%, with a median Duration of Response of 16.7 months. Median PFS was 9.3 months, and median OS was 22.7 months after nearly four years of follow-up. The estimated 4-year OS rate in this cohort was 31%.
Post hoc subgroup analyses suggested that clinical benefit was generally consistent across baseline characteristics. Notably, both PFS and OS were numerically longer in patients without a smoking history compared with those with a history of smoking, a finding that may be related to pharmacokinetic effects on Binimetinib exposure and warrants further investigation.
The safety profile observed with extended follow-up was consistent with prior analyses and with known toxicities associated with BRAF and MEK inhibition. Most treatment-related adverse events were low grade and manageable, with gastrointestinal symptoms, fatigue, and nausea among the most frequently reported. Rates of dose modification and discontinuation were similar across treatment lines, and no new safety signals emerged with longer-term exposure.
Although cross-trial comparisons should be interpreted cautiously, the Overall Survival outcomes observed in PHAROS compare favorably with historical data for targeted therapy in this population. Given that a significant proportion of patients with metastatic NSCLC may not receive subsequent lines of therapy, these findings emphasize the importance of selecting the most effective treatment upfront.
In conclusion, updated results from the PHAROS study demonstrate durable responses and sustained long-term survival with Encorafenib plus Binimetinib in patients with BRAF V600E–mutant mNSCLC. The depth and durability of benefit, particularly in treatment-naïve patients, further support this combination as a preferred first-line targeted therapy option and reinforce its role in the evolving treatment landscape for this molecularly defined NSCLC subgroup.
Updated Overall Survival Analysis From the Phase II PHAROS Study of Encorafenib Plus Binimetinib in Patients With BRAF V600E-Mutant Metastatic Non–Small Cell Lung Cancer. Johnson ML, Smit EF, Felip E, et al. J Clin Oncol. 2025;43:3706-3713