
The FDA on May 18, 2017 granted regular approval to KEYTRUDA® for patients with locally advanced or metastatic Urothelial Carcinoma, who have disease progression during or following Platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with Platinum-containing chemotherapy. KEYTRUDA® is a product of Merck and Co., Inc.
Month: July 2017
KEYTRUDA® (Pembrolizumab)
The FDA on May 10, 2017 granted accelerated approval to KEYTRUDA® in combination with ALIMTA® (Pemetrexed) and Carboplatin for the treatment of patients with previously untreated metastatic Non-Squamous Non-Small Cell Lung Cancer (NSCLC). KEYTRUDA® is a product of Merck and Co., Inc.
FDA Approves TAFINLAR® and MEKINIST® Combination for BRAF Positive Non Small Cell Lung Cancer
SUMMARY: The FDA on June 22, 2017, granted regular approvals to TAFINLAR® (Dabrafenib) and MEKINIST® (Trametinib) administered in combination, for patients with metastatic Non Small Cell Lung Cancer (NSCLC), with BRAF V600E mutation, as detected by an FDA-approved test. These are the first FDA approvals specifically for treatment of patients with BRAF V600E mutation-positive metastatic NSCLC.
The FDA also approved the Oncomine® Dx Target Test, a next generation sequencing (NGS) test to detect multiple gene mutations for lung cancer in a single test from a single tissue specimen. This test detects the presence of BRAF, ROS1, and EGFR gene mutations or alterations in tumor tissue of patients with NSCLC. This test can be used to select patients with NSCLC with the BRAF V600E mutation for treatment with the combination of TAFINLAR® and MEKINIST®. This is the first NGS oncology panel test approved by the FDA for multiple companion diagnostic indications.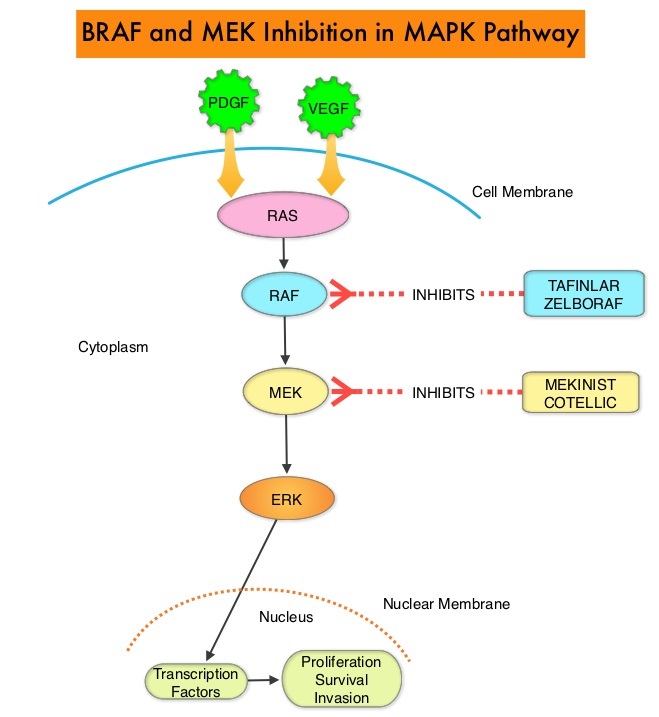
Combining MEKINIST® (Trametinib) with TAFINLAR® (Dabrafenib) to treat patients with NSCLC, was based on the understanding of the biological pathways of this malignancy. The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. This pathway has been implicated in the development of multiple malignancies including NSCLC and Melanoma. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been demonstrated in 6-8% of all malignancies. TAFINLAR® is a selective oral BRAF inhibitor and MEKINIST® is a potent and selective inhibitor of MEK gene, which is downstream from RAF in the MAPK pathway.
The approval of TAFINLAR® and MEKINIST® combination, for patients with metastatic NSCLC was based on an international, multicenter, three-cohort, non-randomized, open-label trial, in patients with locally confirmed BRAF V600E mutation-positive, metastatic NSCLC. In this phase II trial, 93 patients were treated with the combination of TAFINLAR® 150 mg orally twice daily and MEKINIST® 2 mg orally once daily. Of these 93 patients, 36 patients had received no prior systemic therapy for metastatic NSCLC and 57 patients received at least one prior platinum-based chemotherapy regimen and had disease progression. The third cohort in this phase II trial included 78 previously treated patients with BRAF V600E mutation-positive metastatic NSCLC, who received single-agent TAFINLAR®. The primary endpoint was Overall Response Rate (ORR).
It was noted that in the previously treated group, the ORR for the combination treatment based on independent review was 63% with a median Duration of Response of 12.6 months. In the treatment-naive group, the ORR for the combination was 61% and this group had not reached the endpoint for median Duration of Response and therefore was not estimable. However, among those who responded to treatment, 59% of the responders had response durations greater than 6 months. The ORR for patients who received single agent TAFINLAR® was 27% and the median Duration of Response was 9.9 months. The most common Grade 3-4 adverse reactions were pyrexia, fatigue, dyspnea, vomiting, rash, hemorrhage, and diarrhea.
It was concluded that TAFINLAR® plus MEKINIST® combination represents a new targeted therapy for patients with BRAF V600E mutation¬-positive metastatic NSCLC, who tend to respond less favorably to standard chemotherapy. This approval marks the fourth actionable genomic biomarker in metastatic NSCLC along with EGFR, ALK and ROS-1. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Planchard D, Besse B, Groen HJ et al. Lancet Oncol. 2016 Jul;17(7):984-93. doi: 10.1016/S1470-2045(16)30146-2. Epub 2016 Jun 6.
FDA Approves Subcutaneous RITUXAN® Formulation for CD20-Positive Hematologic Malignancies
SUMMARY: The US FDA on June 22, 2017, granted regular approval to the combination of RITUXAN® (Rituximab) and Hyaluronidase human (RITUXAN HYCELA®) for adult patients with Follicular Lymphoma, Diffuse Large B-Cell Lymphoma, and Chronic Lymphocytic Leukemia. RITUXAN HYCELA® is a combination of RITUXAN® and Hyaluronidase human (ENHANZE® technology), for SubCutaneous (SC) injection in multiple hematological malignancies. ENHANZE® is a drug delivery technology platform which removes limitations on the volume of biologics and drugs that can be delivered SubCutaneously, thereby significantly reducing the time required for drug administration.
RITUXAN® is a first generation type I, chimeric, monoclonal antibody that targets the CD20 antigen expressed on the surface of pre-B and mature B-lymphocytes. Upon binding to CD20, RITUXAN® mediates B-cell lysis. Possible mechanisms of cell lysis include Complement Dependent Cytotoxicity (CDC) and Antibody Dependent Cell mediated Cytotoxicity (ADCC).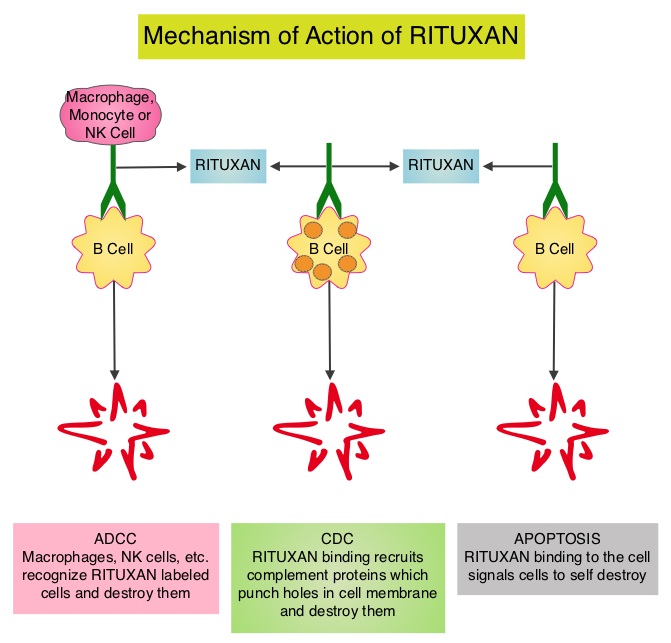 Hyaluronan is a polysaccharide present in the extracellular matrix of the subcutaneous tissue and is depolymerized by the naturally occurring enzyme Hyaluronidase. Hyaluronidase human increases permeability of the subcutaneous tissue locally for a period of 24-48 hrs, by temporarily depolymerizing Hyaluronan.
Hyaluronan is a polysaccharide present in the extracellular matrix of the subcutaneous tissue and is depolymerized by the naturally occurring enzyme Hyaluronidase. Hyaluronidase human increases permeability of the subcutaneous tissue locally for a period of 24-48 hrs, by temporarily depolymerizing Hyaluronan.
The approval of RITUXAN HYCELA® was based on several randomized clinical trials that demonstrated Non-inferior pharmacokinetics of SubCutaneous RITUXAN HYCELA® compared with IV RITUXAN® as well as comparable efficacy and safety results. The following are the specific trials included in the clinical development program:
MabEase phase III trial is an open label study which evaluated SubCutaneous (N=381) versus IV (N=195) RITUXAN® formulation plus CHOP chemotherapy, in treatment naïve patients with CD20-positive Diffuse Large B-Cell Lymphoma. There was no significant difference noted in the Objective Response Rate, Complete Response rates, Progression Free Survival and Overall Survival, between the two treatment groups.
SABRINA is a randomized phase III study that enrolled a total of 410 patients with previously untreated, CD20-positive Follicular Lymphoma and patients were randomized (1:1) to receive either a SC or IV RITUXAN® formulation in combination with CHOP or CVP chemotherapy. There was again no significant difference noted in the Objective Response Rate, Complete Response rates, Progression Free Survival and Overall Survival, between the two treatment groups
The SAWYER study is a phase Ib open label trial which compared the SC and IV formulations of RITUXAN® in combination with Fludarabine and Cyclophosphamide chemotherapy, in 176 treatment naïve patients with Chronic Lymphocytic Leukemia. There was no significant difference noted in the Objective Response Rate, Complete Response rates, Progression Free Survival and Overall Survival, between the two treatment groups.
SparkThera is a phase Ib study which investigated the pharmacokinetics and safety of SubCutaneous (SC) versus IV RITUXAN® formulations as maintenance therapy, in previously untreated or relapsed Follicular Lymphoma. It was noted that the pharmacokinetics of SC RITUXAN® formulation was Non-inferior to IV RITUXAN® formulation, with a comparable safety profile.
PrefMab is a randomized, open label, phase IIIb study which evaluated patient preference for SC or IV RITUXAN® formulation, in previously untreated CD20-positive Follicular lymphoma and Diffuse Large B-Cell Lymphoma. In this study, most patients preferred SC compared with IV formulation of RITUXAN®, mainly due to reduction in the duration and discomfort of administration.
Treatment with RITUXAN HYCELA® should be initiated only after patients had received at least one full dose RITUXAN® by intravenous infusion. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761064s000lbl.pdf
FDA Approves Subcutaneous RITUXAN® Formulation for CD20-Positive Hematologic Malignancies
The US FDA on June 22, 2017, granted regular approval to the combination of RITUXAN® (Rituximab) and Hyaluronidase human (RITUXAN HYCELA®) for adult patients with Follicular Lymphoma, Diffuse Large B-Cell Lymphoma, and Chronic Lymphocytic Leukemia. This Subcutaneous formulation of RITUXAN® utilizes ENHANZE®, a drug delivery technology platform, which removes limitations on the volume of biologics and drugs that can be delivered Subcutaneously, thereby significantly reducing the time required for drug administration. The approval of RITUXAN HYCELA® was based on several randomized clinical trials that demonstrated Non-inferior pharmacokinetics of Subcutaneous RITUXAN HYCELA® compared with IV RITUXAN®, as well as comparable efficacy and safety results.