⇚
 Advertisement
Advertisement Lung Cancer: Non-Small Cell
FDA Grants Regular Approval to TABRECTA® for Metastatic Non-Small Cell Lung Cancer
SUMMARY: The FDA on August 10, 2022, granted regular approval to TABRECTA® (Capmatinib), for adult patients with metastatic Non-Small Cell Lung Cancer (NSCLC) whose tumors have a mutation leading to Mesenchymal-Epithelial Transition (MET) exon 14 skipping, as detected by an FDA-approved test.

The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
MET is a widely expressed Receptor Tyrosine Kinase and plays a pivotal role in cell growth, proliferation, and survival. The MET gene encodes for a protein known as the Hepatocyte Growth Factor (HGF) Receptor. Upon binding by Hepatocyte Growth Factor (HGF), the HGF Receptor is activated, with resulting activation of the downstream RAS/RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways, thereby serving different important biological functions. Alterations in the MET gene leading to abnormal MET signaling, has been identified in different types of cancers including thyroid, lung, breast, liver, colon, kidney, ovary, and gastric carcinoma.
Two key MET alterations include MET exon 14 skipping mutations and MET amplification. MET exon 14 skipping mutations occur in approximately 5% of NSCLC patients with enrichment in sarcomatoid lung cancers (22%). MET exon 14 skipping mutation is a recognized oncogenic driver and is a molecular genetic abnormality indicating the presence of a splice site mutation that results in a loss of transcription of exon 14 of the MET gene. Most exon 14 mutations occur in never-smokers and is seen in both squamous and adenocarcinoma histology. Patients whose cancers have MET exon 14 skipping generally have very high response rates to MET inhibitors and molecular testing for MET exon 14 skipping should therefore be performed on all lung cancers, because this is a targetable alteration. MET amplification has been more commonly seen in smokers, and responses in patients with MET-amplified tumors might be more variable and dependent on level of amplification, with higher responses noted in tumors with more than 5-6- fold amplification. Tumors with MET exon 14 skipping mutations usually do not harbor activating mutations in EGFR, KRAS, or BRAF or concurrent ALK, ROS1 or RET translocations. However, it appears that cMET exon 14 skipping is not mutually exclusive with cMET amplification.
TABRECTA® is a highly potent and selective, reversible inhibitor of MET tyrosine kinase. The FDA in May 2020 granted accelerated approval for the same indication based on the primary findings from the GEOMETRY mono-1 trial, which is a non-randomized, open-label, multi-cohort, Phase II study, conducted to evaluate the efficacy and safety of single-agent TABRECTA® in adult patients with EGFR wild-type, ALK-negative, metastatic NSCLC, whose tumors have a mutation that leads to MET exon 14 skipping (METex14), as detected by an RNA-based RT-PCR. The conversion to regular approval was based on data from an additional 63 patients (Total N=160), as well as an additional 22 months of follow- up time, to assess durability of response and verify clinical benefit.
In this updated analysis, a total of 160 patients (N=160) with metastatic NSCLC and confirmed MET exon 14 skipping mutations were included, of whom 60 patients were treatment naïve and 100 patients were previously treated. The patients received TABRECTA® at 400 mg orally twice daily until disease progression or unacceptable toxicity. The median patient age was 71 years and all NSCLC histologies including sarcomatoid/carcinosarcoma were included. Majority of the patients (77%) were white and 23% were Asian, 61% never smoked, 83% had adenocarcinoma, and 16% had CNS metastases. Among previously treated patients, 81% received one, 16% received two, and 3% received three prior lines of systemic therapy. Amongst previously treated patients, 86% received prior platinum-based chemotherapy. The Primary efficacy outcome was Overall Response Rate (ORR), and additional efficacy outcomes included Duration of Response, Time to Response, Disease Control Rate, Progression Free Survival (PFS) and Safety, as determined by a Blinded Independent Review Committee (BIRC).
Among the treatment-naïve patients (N=60), the ORR was 68% with a median Duration of Response of 12.6 months. Among the previously treated patients (N=100), the ORR was 44%, with a median Duration of Response of 9.7 months. The most common adverse events (occurring in at least 20% of patients) were peripheral edema, nausea, fatigue, vomiting, dyspnea, and decreased appetite. TABRECTA® can also cause Interstitial Lung Disease, hepatotoxicity and photosensitivity.
It was concluded that TABRECTA® is a new treatment option for patients with MET exon 14 skipping- mutated advanced NSCLC, regardless of the line of therapy, with deep and durable responses, and with manageable toxicity profile.
Capmatinib in MET exon 14-mutated, advanced NSCLC: Updated results from the GEOMETRY mono-1 study. Wolf J, Garon EB, Groen HJM, et al. DOI: 10.1200/JCO.2021.39.15_suppl.9020 Journal of Clinical Oncology - published online before print May 28, 2021.
FDA Grants Accelerated Approval to ENHERTU® for HER2-Mutant Non Small Cell Lung Cancer
SUMMARY: The FDA on August 11, 2022, granted accelerated approval to ENHERTU® (fam-trastuzumab deruxtecan-nxki), for adult patients with unresectable or metastatic Non-Small Cell Lung Cancer (NSCLC) whose tumors have activating human Epidermal Growth Factor Receptor 2 or HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have received a prior systemic therapy. This is the first drug approved for HER2-mutant NSCLC. FDA also approved Oncomine™ Dx Target Test (tissue) and Guardant360® CDx (plasma) as companion diagnostics for ENHERTU®. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. HER2 is a Tyrosine Kinase Receptor expressed on the surface of several tumor types including Breast, Gastric, Lung and Colorectal cancers. It is a growth-promoting protein, and HER2 overexpression/HER2 gene amplification is often associated with aggressive disease and poor prognosis in certain tumor types. However, HER2 overexpression and gene amplification are associated with distinct molecular entities and have limited therapeutic value in lung cancer.
HER2 mutations unlike HER2 overexpression and gene amplification are oncogenic drivers and are detected in 2 to 4% of NSCLCs. They are more often detected in younger, female and never-smokers, and almost exclusively in Adenocarcinomas. Next-generation sequencing is used to identify HER2 mutations. Majority of HER2 mutations (80-90%) occur in exon 20, as either a duplication or an insertion of 12 nucleotides, resulting in the addition of four amino acids (YVMA) at codon 775 in the kinase domain. This distinct molecular entity is characterized by specific pathological and clinical behavior. These acquired HER2 gene mutations have been independently associated with cancer cell growth, aggressive form of disease and poor prognosis, and with an increased incidence of brain metastases. There are currently no therapies approved specifically for the treatment HER2 mutant NSCLC and is therefore an unmet need.
ENHERTU® (Trastuzumab Deruxtecan) is an Antibody-Drug Conjugate (ADC) composed of a humanized monoclonal antibody specifically targeting HER2, with the amino acid sequence similar to HERCEPTIN® (Trastuzumab), attached to a potent cytotoxic Topoisomerase I inhibitor payload by a cleavable tetrapeptide-based linker. ENHERTU® has a favorable pharmacokinetic profile and the tetrapeptide-based linker is stable in the plasma and is selectively cleaved by cathepsins that are up-regulated in tumor cells. Unlike KADCYLA® (ado-Trastuzumab emtansine), which is also an Antibody-Drug Conjugate, ENHERTU® has a higher drug-to-antibody ratio (8 versus 4), the released payload easily crosses the cell membrane with resulting potent cytotoxic effect on neighboring tumor cells regardless of target expression, and the released cytotoxic agent (payload) has a short half-life, minimizing systemic exposure. ENHERTU® is approved in the US for the treatment of adult patients with unresectable or metastatic HER2-positive or HER2-Low breast cancer and locally advanced or metastatic HER2-positive Gastric or GastroEsophageal Junction adenocarcinoma who have received a prior Trastuzumab based regimen. Translational research demonstrated that HER2-mutant NSCLC may preferentially internalize the HER2 receptor Antibody-Drug Conjugate complex regardless of HER2 protein expression and overcome resistance to other HER2-targeted agents.
In the DESTINY-Lung01 Phase II, open-label, two-cohort trial of heavily pretreated population of patients with HER2-mutated advanced NSCLC, treatment with ENHERTU® 6.4 mg/kg given by IV infusion every 3 weeks resulted in an Objective Response Rate (ORR) of 55%, with a median Duration of Response was 9.3 months. Responses were observed across different HER2 mutation subtypes. The median PFS was 8.2 months, and the median OS was 17.8 months (NEJM 2022;386:241-251).
The present FDA approval was based on DESTINY-Lung02, which is a global, multicenter, multi-cohort, randomized, blinded, dose-optimization, Phase II trial, in which the safety and efficacy of two doses ENHERTU® (5.4mg/kg or 6.4mg/kg) was evaluated, in patients with HER2 mutated metastatic NSCLC, with disease recurrence or progression during or after at least one regimen of prior anticancer therapy that must have contained a platinum-based chemotherapy. This study enrolled 152 patients (N=152) and patients were selected for treatment with ENHERTU® based on the presence of activating HER2 (ERBB2) mutations in a tumor specimen. Patients were randomized to receive ENHERTU® 6.4 mg/kg or 5.4 mg/kg by IV infusion every 3 weeks, until unacceptable toxicity or disease progression. The Primary endpoint of the trial was Objective Response Rate (ORR) as assessed by Blinded Independent Central Review (BICR). Secondary endpoints included Disease Control Rate (DCR), Duration of Response (DoR), Progression Free Survival (PFS), Overall Survival (OS) and Safety. The primary/interim efficacy analysis included a pre-specified cohort of 52 patients (N=52). The median age in this cohort was 58 years, 69% were female; 79% were Asian, 12% were White, and 10% were of other races.
ENHERTU® 5.4mg/kg IV demonstrated a confirmed Objective Response Rate of 57.7%, with a Complete Response Rate of 1.9%, Partial Response Rate of 55.8%, and median Duration of Response of 8.7 months. The most common adverse effects included nausea, alopecia, increased AST and ALT, cytopenias, and was consistent with previous clinical trials, with no new safety concerns identified.
It was concluded that ENHERTU® is the first HER2-directed treatment option for patients with HER2 mutated NSCLC, and fulfills an unmet medical need in this patient population.
https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-fam-trastuzumab-deruxtecan-nxki-her2-mutant-non-small-cell-lung
Late Breaking Abstract – ASCO 2022: Landmark Five Year Overall Survival Rates for OPDIVO® and YERVOY® Combination in NSCLC
SUMMARY: The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the immune system T cells. Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152) and PD-1(Programmed cell Death 1). Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response. OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4.
CheckMate-227 is an open-label, multi-part, global, Phase III trial in which OPDIVO® based regimens were compared with Platinum-doublet chemotherapy in patients with first line advanced NSCLC, across non-squamous and squamous tumor histologies. This study consisted of Part 1a/Part 1b and Part 2. In Part 2 of this trial, OPDIVO® plus chemotherapy was compared with chemotherapy alone, regardless of PD-L1 expression. Part 2 did not meet its Primary endpoint for Overall Survival for OPDIVO® plus chemotherapy versus chemotherapy alone, in patients with non-squamous NSCLC, and is published elsewhere.
Part 1a: Patients received OPDIVO® 3 mg/kg IV every 2 weeks plus YERVOY® 1 mg/kg IV every 6 weeks (N=396), OPDIVO® monotherapy 240 mg IV every 2 weeks (N=396) or chemotherapy alone given every 3 weeks for up to four cycles (N=397), in patients whose tumors had PD-L1 expression of 1% or more.
Part 1b: Patients received OPDIVO® plus YERVOY® (N=187), OPDIVO® 360 mg IV every 3 weeks plus chemotherapy IV every 3 weeks for up to four cycles (N=177), or chemotherapy alone IV every 3 weeks for up to four cycles (N=186), in patients whose tumors did not express PD-L1 (less than 1%)
Patients were stratified by histology, and treatment was administered until disease progression, unacceptable toxicity, or administered for 2 years for immunotherapy. It should be noted that when this trial was launched, chemotherapy along with immunotherapy or immunotherapy alone was not approved for the front-line treatment of NSCLC. Therefore, dual immunotherapy combination was not compared with current standards of care such as chemotherapy plus immunotherapy.
There were two independent Primary endpoints in Part 1 for OPDIVO® plus YERVOY® versus chemotherapy: Overall survival (OS) in patients whose tumors express PD-L1 (assessed in patients enrolled in Part 1a) and Progression Free Survival (PFS) in patients with TMB of 10 mut/Mb or more, across the PD-L1 spectrum (assessed in patients enrolled across Part 1a and Part 1b). Other assessments included Objective Response Rate (ORR), Duration of Response (DOR), and treatment-free interval. Treatment-free interval was measured in patients who discontinued study therapy and was defined as the time from last study dose to start of subsequent systemic therapy.
The Overall Survival (OS) data was previously reported at a minimum follow up of 29 months, and the median OS was of 17.1 months for the OPDIVO® plus YERVOY® group, compared to 14.9 months in the chemotherapy group (HR=0.79; P=0.007), with a 2-year OS rate of 40.0% and 32.8%, respectively. The researchers here in presented data after a minimum follow up of 61.3 months (5 years).
Patients whose tumors had PD-L1 expression of 1% or more continued to have sustained long term OS benefit with OPDIVO® plus YERVOY® when compared to chemotherapy (HR=0.77), and the 5-year OS rates were 24% with OPDIVO® plus YERVOY® compared to 14% with chemotherapy alone.
Patients with a PD-L1 expression of less than 1% also demonstrated continued long term OS benefit with OPDIVO® plus YERVOY® when compared to chemotherapy (HR = 0.65), and the 5-year OS rates were 19% for OPDIVO® plus YERVOY® compared to 7% for chemotherapy alone.
Among patients who survived for 5 years, median PFS was 59.1 months for PD-L1–positive patients and 60.7 months for PD-L1–negative patients who received OPDIVO® plus YERVOY®, compared to 9.5 months and 24.9 months respectively, for those who received chemotherapy.
Among those who responded to treatment, more patients who received OPDIVO® plus YERVOY® remained in response at five years, compared to chemotherapy, in both PD-L1 expression of 1% or more group (28% versus 3%) and PD-L1 expression of less than 1% group (21% versus 0%), respectively.
Among patients treated with OPDIVO® plus YERVOY® who were alive at five years, approximately two-thirds of patients did not receive any subsequent therapy for more than three years after stopping treatment, regardless of PD-L1 expression.
It was concluded that in this longest reported follow up of a Phase III trial of first line, chemotherapy free, combination immunotherapy, in metastatic Non Small cell Lung Cancer, a combination of OPDIVO® plus YERVOY® continued to provide long term durable clinical benefit and increased 5-year survivorship, when compared to chemotherapy, in previously untreated patients with metastatic NSCLC, regardless of PD-L1 expression.
Five-year survival outcomes with nivolumab (NIVO) plus ipilimumab (IPI) versus chemotherapy (chemo) as first-line (1L) treatment for metastatic non–small cell lung cancer (NSCLC): Results from CheckMate 227. Brahmer JR, Lee J-S, Ciuleanu T-E, et al. J Clin Oncol. 2022;40(suppl 17):LBA9025. doi:10.1200/JCO.2022.40.17_suppl.LBA9025
Late Breaking Abstract – ASCO 2022: Adagrasib in KRAS G12C Mutated Non Small Cell Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The KRAS (kirsten rat sarcoma viral oncogene homologue) proto-oncogene encodes a protein that is a member of the small GTPase super family. The KRAS gene provides instructions for making the KRAS protein, which is a part of a signaling pathway known as the RAS/MAPK pathway. By relaying signals from outside the cell to the cell nucleus, the protein instructs the cell to grow, divide and differentiate. The KRAS protein is a GTPase, and converts GTP into GDP. To transmit signals, the KRAS protein must be turned on by binding to a molecule of GTP. When GTP is converted to GDP, the KRAS protein is turned off or inactivated, and when the KRAS protein is bound to GDP, it does not relay signals to the cell nucleus. The KRAS gene is in the Ras family of oncogenes, which also includes two other genes, HRAS and NRAS. When mutated, oncogenes have the potential to change normal cells cancerous.
KRAS is the most frequently mutated oncogene in human cancers and are often associated with resistance to targeted therapies and poor outcomes. The KRAS G12C mutation occurs in approximately 25% of Non Small Cell Lung Cancers (NSCLC) and in 3-5% of colorectal cancers and other solid cancers. KRAS G12C is one of the most prevalent driver mutations in NSCLC and accounts for a greater number of patients than those with ALK, ROS1, RET, and TRK 1/2/3 mutations combined. KRAS G12C cancers are genomically more heterogeneous and occur more frequently in current or former smokers, and are likely to be more complex genomically than EGFR mutant or ALK rearranged cancers. G12C is a single point mutation with a Glycine-to-Cysteine substitution at codon 12. This substitution favors the activated state of KRAS, resulting in a predominantly GTP-bound KRAS oncoprotein, amplifying signaling pathways that lead to oncogenesis.
Adagrasib is a potent, orally available, small molecule covalent inhibitor of KRAS G12C. This drug irreversibly and selectively binds KRAS G12C in its inactive, GDP-bound state. Unlike LUMAKRAS® (Sotorasib), which is also a selective covalent inhibitor of KRAS G12C, Adagrasib has a longer drug half-life of 23 hours, as compared to 5 hours for LUMAKRAS®, has dose-dependent extended exposure, and can penetrate the CNS. Approximately, 27-42% of patients with NSCLC harboring KRAS G12C mutations have CNS metastases, with poor outcomes.
KRYSTAL-1 is a Phase I/II multiple expansion cohort trial involving patients with advanced solid tumors harboring a KRAS G12C mutation. Adagrasib demonstrated clinical activity in patients with KRAS G12C-mutated solid tumors, including colorectal, pancreatic, and biliary tract cancers. Further, preliminary data from two patients with untreated CNS metastases from a Phase 1b cohort showed antitumor activity against CNS metastases, with satisfactory concentrations of Adagrasib in the CSF.
The researchers in this publication reported the results from Cohort A, a Phase 2 cohort of the KRYSTAL-1 study in which Adagrasib at a dose of 600 mg orally twice daily was evaluated in patients with KRAS G12C-mutated NSCLC, previously treated with chemotherapy and anti-Programmed Death 1 (PD-1) or Programmed Death Ligand 1 (PD-L1) therapy. This registration study included a total of 116 unresectable or metastatic NSCLC patients, with histologically confirmed diagnosis, with KRAS G12C mutation (detected in tumor tissue at a local or central laboratory), who had previously received treatment with at least one platinum-containing chemotherapy regimen and checkpoint inhibitor therapy (in sequence or concurrently), and who had measurable tumor lesions. Enrolled patients received Adagrasib 600 mg capsule twice daily, and treatment was continued until disease progression or unacceptable toxicities. The median patient age was 64 years, 97% had adenocarcinoma histology, 98% had both platinum based therapy and checkpoint inhibitor therapy, and 21% of patients had CNS metastases. Key exclusion criteria included active CNS metastases (patients were eligible if CNS metastases were adequately treated and neurologically stable), carcinomatous meningitis, and previous treatment with a KRAS G12C inhibitor. Exploratory Biomarker Analyses included candidate biomarkers (PD-L1 Tumor Proportion Score and mutational status of STK11, KEAP1, TP53, and CDKN2A on tumor-tissue specimens, blood specimens, or both, for their association with tumor response. The Primary end point was Objective Response Rate as assessed by blinded Independent Central Review. Secondary end points included the Duration of Response, Progression Free Survival, Overall Survival, and safety.
The median follow up was 12.9 months and the median duration of treatment was 5.7 months. The confirmed Objective Response Rate was 42.9% and the median Duration of Response was 8.5 months. The median Progression Free Survival was 6.5 months and the median Overall Survival was 12.6 months, at a median follow up of 15.6 months. Among 33 patients with previously treated, stable CNS metastases, the intracranial confirmed Objective Response Rate was 33.3%. Treatment-related adverse events occurred in 97.4% of the patients and 53% were Grade 1 or 2 toxicities. Adagrasib was discontinued in 6.9% of patients due to adverse events.
It was concluded that among patients with previously treated KRAS G12C-mutated NSCLC, Adagrasib showed significant clinical efficacy without new safety signals, and encouraging intracranial activity. The researchers added that these are the first clinical data demonstrating CNS-specific activity of a KRAS G12C inhibitor in this patient population.
Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. Jänne PA, Riely GJ, Gadgeel SM, et al. DOI: 10.1056/NEJMoa2204619
Consider Guideline-Recommended Biomarker Testing as an Integral Component of NSCLC Care
The NSCLC Landscape Has Evolved Significantly Due Largely to the Growing Number of Actionable Mutations1
Despite advancements in standard-of-care, advanced non-small cell lung cancer (NSCLC) continues to burden patients, with poor survival outcomes.2,3 NSCLC has been identified as the leading cause of cancer death worldwide with an estimated 1.8 million deaths in 2020.2 As the number of targeted therapies and approved companion diagnostics continues to grow, mortality and survival rates have begun to improve.3 With the addition of KRAS G12C, there are 9 actionable molecular biomarkers (as of February 2022) and more than 20 targeted therapies approved for use in advanced NSCLC.1,4 Guidelines recommend biomarker testing for all eligible patients at diagnosis of advanced NSCLC regardless of characteristics such as smoking history, race, or histology.5,6 Unfortunately, real-world evidence shows that far too many patients fail to receive the comprehensive biomarker testing.7
Adherence to Guidelines Can Improve Patient Outcomes8
As targeted therapies are approved, guidelines continue to update their recommendations on biomarker testing.5 As of March 2022, NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC recommend broad molecular testing of actionable and emerging biomarkers for eligible patients with advanced or metastatic NSCLC (Figure 1).5 Similarly, the American Society of Clinical Oncology (ASCO) endorsed the 2018 College of American Pathologists (CAP)/International Association for the Study of Lung Cancer (IASLC)/Association for Molecular Pathology (AMP) guidelines, recommending comprehensive cancer panel testing for genetic biomarkers.9,10
Figure 1: NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC5,*,†
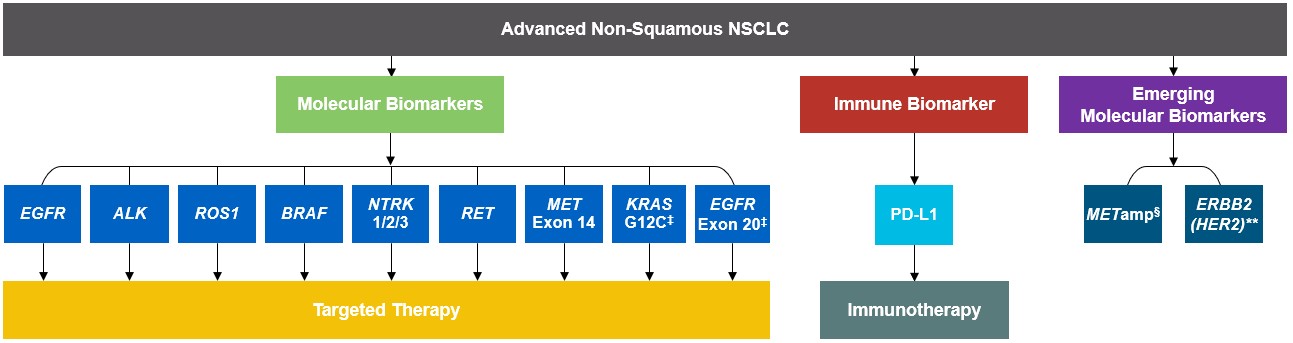
*The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC provide recommendations for certain individual biomarkers that should be tested and recommend testing techniques but do not endorse any specific commercially available biomarker assays or commercial laboratories.5†The NCCN Guidelines® for NSCLC recommend broad molecular testing to identify rare driver variants for which targeted therapies may be available to ensure patients receive the most appropriate treatment.5‡KRAS G12C and EGFR exon 20 mutations are used to determine subsequent (ie, second-line and beyond) therapy using targeted agents or other novel agents.5 §The definition of high-level MET amplification is evolving and may differ according to the assay used for testing. For NGS-based results, a copy number greater than 10 is consistent with high-level MET amplification.5 **For oncogenic or likely oncogenic HER2 mutations, refer to definitions at oncokb.org.5
Although adherence to guideline-recommended biomarker testing is associated with improved patient outcomes, real-world EMR data reveals suboptimal biomarker testing rates.8,11 In a retrospective study,†† 81% of patients with metastatic NSCLC did not receive testing for ALK, EGFR, ROS1, and BRAF before initiation of first-line treatment, despite availability of targeted therapies.11 Moreover, only 28% of patients received testing for all four genetic biomarkers and PD-L1 during the study period.11 In another retrospective study, less than 50% of patients with metastatic NSCLC received testing for all five biomarkers (EGFR, ALK, ROS1, BRAF, PD-L1) (Figure 2).7
Beyond the underutilization of biomarker testing, there remains an even greater need to increase broad molecular testing among racial and ethnic minority groups in the US.12,13 In one retrospective study, Black/African American patients with advanced NSCLC had significantly lower rates of testing with NGS assays (39.8%) compared with White patients (50.1%) (Figure 3).12
††A retrospective study assessing real-world biomarker testing patterns in patients with de novo mNSCLC (N=2,257) in the community oncology setting using the US Oncology Network electronic health records between January 1st, 2017 and September 31st, 2019 with follow-up through December 31st, 2019.11
Figure 2: MYLUNG Consortium™ EMR Analysis of Patients With Metastatic NSCLC7,‡‡
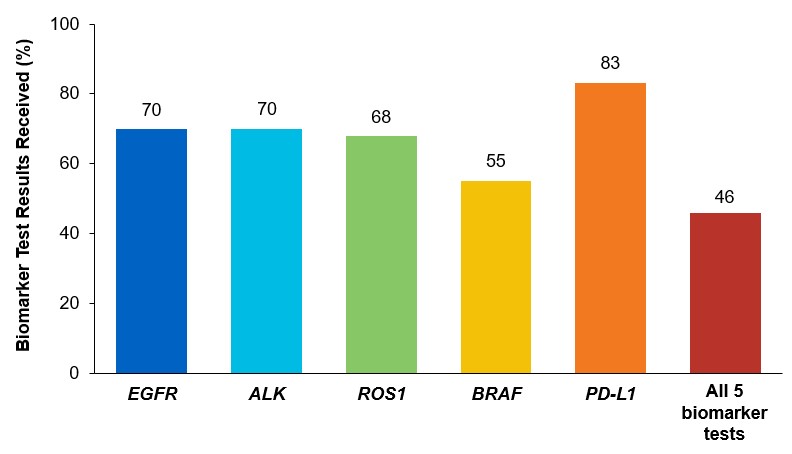
‡‡A retrospective, observational study assessing real-world biomarker testing patterns in patients with metastatic NSCLC(N=3,474) from community oncology practices within the US Oncology Network community practices between 2018 and 2020.7
Figure 3: EMR Analysis of Biomarker Testing in Patients With Advanced/Metastatic NSCLC12,§§
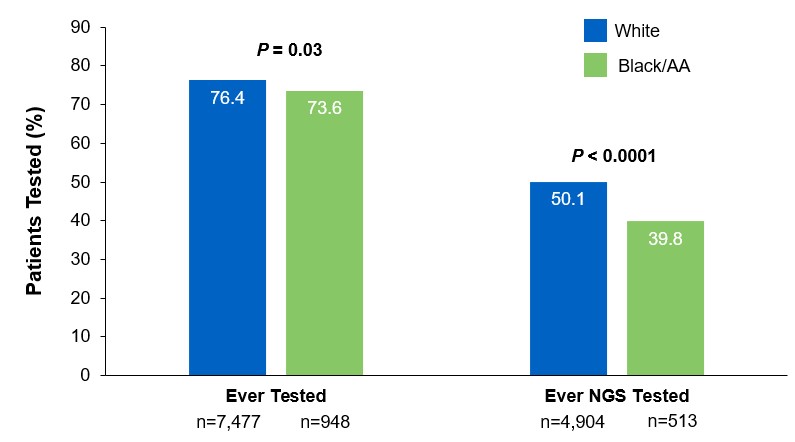
§§From a retrospective cohort study of patients with advanced/metastatic: NSCLC (N=14,768) from ~800 sites of care identified via the Flatiron Electronic Health Record Database between 2017 and 2020. Of this study cohort, patients included White (n=9,793), Black/African American (n=1,288), and non-squamous NSCLC (n=10,333).
Collectively, these findings highlight the disparity in proactive disease management across different patient populations.7,11,12
Considerations Across the Biomarker Testing Journey
There are several different methods in which eligible patients can be tested for actionable genetic alterations, each with unique considerations as indicated below (Figure 4).
Figure 4: Comparing Biomarker Testing Methods and Sample Types
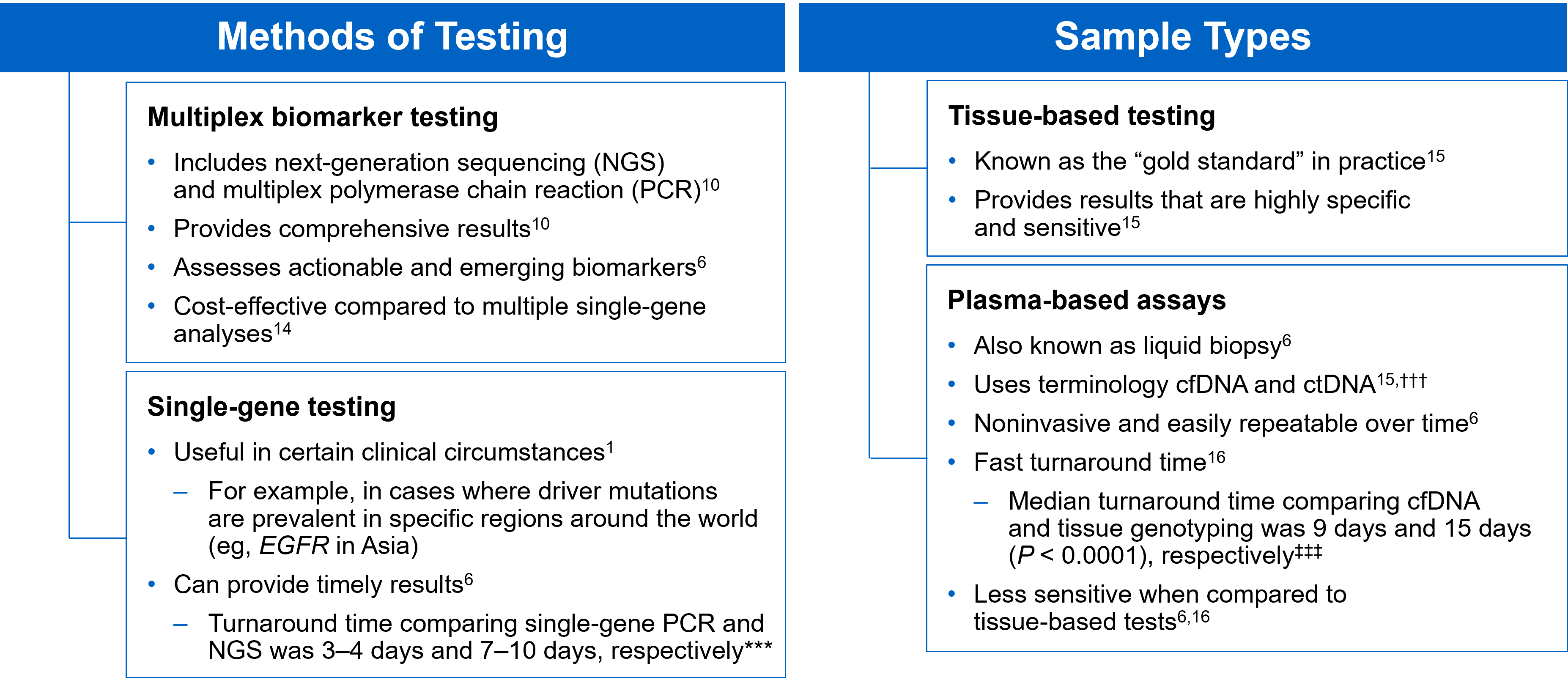
***Data from a review of common molecular assays for biomarker testing that analyzed common detected variants, sensitivities, and turnaround time.6 †††cfDNA refers to all circulating DNA (largely non-malignant), while ctDNA refers to the tumor-related component of cfDNA.15 ‡‡‡Data from a prospective study that enrolled patients with previously untreated metastatic NSCLC undergoing SOC tissue genotyping and comprehensive cfDNA analysis, with turnaround time defined as the number of days between test order date and the retrieval of test results.16
While tissue biopsy remains the “gold standard” in NSCLC, it may not be feasible (insufficient tissue) or pragmatic (urgent need to begin treatment) in all patients.17 Plasma ctDNA demonstrates complementary results to tissue-based assays and can be considered a valid tool for genotyping of newly diagnosed patients with advanced NSCLC.15 In a prospective study of patients with previously untreated, non-squamous metastatic NSCLC from 2016 to 2018, guideline-recommended biomarkers with FDA-approved therapies (EGFR Exon 19 deletion and L858R, ALK fusion, ROS1 fusion, BRAF V600E) showed ≥ 98.2% concordance between tissue and liquid-based testing.16 While concordance is high for any single test, high concordance for full panels will be required for liquid biopsies to become standard; additionally, negative results on liquid biopsy still require validation with tissue testing.16,17
Liquid biopsy may offer improvements in sample acquisition and small tissue samples and provides less invasive procedures and shortened turnaround times.17 Other considerations for maximizing the tissue journey include the use of comprehensive testing, rapid on-site evaluation (ROSE), and implementing reflex testing protocols with the help of a multidisciplinary team (MDT).17
Delays in Biomarker Testing Results May Impact Treatment Plan Decisions18
Longer turnaround times for molecular testing compared with turnaround times for PD-L1 testing by IHC may result in the initiation of immunotherapy before molecular testing results are received.18 Waiting for complete biomarker test results prior to initiating therapy can allow doctors to make the most informed decisions surrounding a patient’s treatment journey.18
Consider Comprehensive Biomarker Testing as an Important Part of Your Treatment Plan8
As the NSCLC landscape continues to progress with the increasing number of actionable biomarkers, there is a growing need for proactive and comprehensive molecular testing.7,17 Although real-world data has shown significant underuse of biomarker testing, rates can be improved with diligent observation of expanding guidelines and recommendations by expert panels and associations.7,8 In the coming years, clinicians may consider evolving institutional protocols, including enabling reflex testing, and work as an MDT to ensure biomarker testing is performed on all eligible patients with advanced NSCLC.17
[Abbreviations]
AA, African American; ALK, anaplastic lymphoma kinase; BRAF, proto-oncogene B-Raf; cfDNA, cell-free DNA; ctDNA, circulating tumor DNA; EGFR, epidermal growth factor receptor; EMR, electronic medical record; ERBB2, erb-b2 receptor tyrosine kinase 2; HER2, human epidermal growth factor receptor 2; IHC, immunohistochemistry; KRAS, Kirsten rat sarcoma viral oncogene homolog; MET, mesenchymal-to-epithelial transition; mNSCLC, metastatic non-small cell lung cancer; NSCLC, non-small cell lung cancer; NCCN, National Comprehensive Cancer Network; NGS, next-generation sequencing; NTRK, neurotrophic tyrosine receptor kinase; PD-L1, programmed cell death ligand 1; RET, rearranged during transfection; ROS1, c-ros oncogene 1; SOC, standard-of-care.
[References]
1. Majeed U, et al. J Hematol Oncol. 2021;14:108.
2. Sung H, et al. CA Cancer J Clin. 2021;71:209-249.
3. Siegel RL, et al. CA Cancer J Clin. 2021;71:7-33.
4. Food and Drug Administration. www.fda.gov. Accessed October 6, 2021.
5. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer. V.3.2022. ©National Comprehensive Cancer Network, Inc. 2022. All rights reserved. Accessed March 16, 2022. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
6. Pennell NA, et al. Am Soc Clin Oncol Educ Book. 2019;39:531-542.
7. Robert NJ, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 102.
8. John A, et al. Adv Ther. 2021;38:1552-1566.
9. Hanna N, et al. J Clin Oncol. 2017;35:3484-3515.
10. Lindeman NI, et al. Arch Pathol Lab Med. 2018;142:321-346.
11. Nadler ES, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 9079.
12. Bruno DS, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 9005.
13. Hann KEJ, et al. BMC Public Health. 2017;17:503.
14. Pennell NA, et al. JCO Precis Oncol. 2019;3:1-9.
15. Rolfo C, et al. J Thorac Oncol. 2021;16:1647-1662.
16. Leighl NB, et al. Clin Cancer Res. 2019;25:4691-4700.
17. Gregg JP, et al. Transl Lung Cancer Res. 2019;8:286-301.
18. Smeltzer MP, et al. J Thorac Oncol. 2020;15:1434-1448.
USA-510-80864 02/22
Mutations of STK11/KRAS Genes and Efficacy of Immunotherapy in NSCLC
SUMMARY: The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers and Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the T cells of the immune system. Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152) and PD-1(Programmed cell Death 1). Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response.
TECENTRIQ® (Atezolizumab) is an anti-PDL1 monoclonal antibody, designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors and thus enabling the activation of T cells. AVASTIN® (Bevacizumab) is a biologic antiangiogenic antibody, directed against Vascular Endothelial Growth Factor (VEGF), and prevents the interaction of VEGF to its receptors (Flt-1 and KDR) on the surface of endothelial cells. The interaction of VEGF with its receptors has been shown to result in endothelial cell proliferation and new blood vessel formation. Combining TECENTRIQ® and AVASTIN® is supported by the following scientific rationale. AVASTIN® in addition to its established anti-angiogenic effects, may further enhance the ability of TECENTRIQ® to restore anti-cancer immunity, by inhibiting VEGF-related immunosuppression, promoting T-cell tumor infiltration and enabling priming and activation of T-cell responses against tumor antigens.
IMpower150 is a multicenter, open-label, randomized, Phase III study, conducted to evaluate the efficacy and safety of TECENTRIQ® in combination with Carboplatin and Paclitaxel with or without AVASTIN®, in patients with Stage IV, treatment naïve, non-squamous NSCLC. This study enrolled 1,202 patients, who were randomized (1:1:1) to receive either TECENTRIQ® along with Carboplatin and Paclitaxel (ACP-Group A), TECENTRIQ® and AVASTIN® along with Carboplatin and Paclitaxel (ABCP-Group B), or AVASTIN® plus Carboplatin and Paclitaxel (BCP-Group C - control arm). During the treatment-induction phase, patients in Group A received TECENTRIQ® 1200 mg IV along with Carboplatin AUC 6 and Paclitaxel 200mg/m2 IV on Day 1 of a 3-week treatment cycle for 4 or 6 cycles. Following the induction phase, patients received maintenance treatment with TECENTRIQ® on the same dose schedule until disease progression. Patients in Group B received AVASTIN® 15 mg/kg IV, along with TECENTRIQ®, Carboplatin and Paclitaxel IV, Day 1 of a 3-week treatment cycle for 4 or 6 cycles followed by maintenance treatment with the TECENTRIQ® and AVASTIN® until disease progression. Patients in the control Group C received AVASTIN® plus Carboplatin and Paclitaxel every 3 weeks for 4 or 6 cycles followed by AVASTIN® maintenance treatment until disease progression. Among randomized patients with tumors demonstrating no ALK and EGFR mutations, ABCP was associated with significant improvements in Progression Free Survival (PFS) and Overall Survival (OS), compared with BCP, in an updated OS analysis. ABCP also prolonged OS and PFS compared with BCP in an exploratory subgroup analysis of patients with EGFR-sensitizing mutations.
The Serine‐Threonine Kinase 11 (STK11) gene is located on the short arm of chromosome 19 and germline STK11 mutations are often detected in Peutz‐Jeghers syndrome, an Autosomal Dominant disorder resulting in mucocutaneous hyperpigmentation, hamartomas throughout the gastrointestinal tract, and a predisposition for breast, lung, pancreas, and gastrointestinal malignancies including cancers of the colon and small bowel. Both STK11 (also called LKB1) and KEAP1 mutation occur in about 17% of NSCLC (adenocarcinomas), respectively, and correlates with poor outcome with immune checkpoint inhibitors or immune checkpoint inhibitors plus chemotherapy. Although immune checkpoint inhibitors with or without chemotherapy have demonstrated survival benefit in patients with KRAS mutated tumors, it remains unclear how co-occurring STK11, KEAP1, and TP53 mutations affect outcomes following immune checkpoint blockade.
The authors in this publication conducted a retrospective exploratory analysis of the efficacy of ABCP (TECENTRIQ® and AVASTIN® along with Carboplatin and Paclitaxel), in patients with KRAS mutations and co-occuring STK11, KEAP1, or TP53 mutations, from the IMpower150 nonsquamous NSCLC patient population. Mutation status was determined by circulating tumor DNA Next-Generation Sequencing.
Among the KRAS mutated population, there was numerical improvement in median OS with ABCP compared to BCP (19.8 vs 9.9 months; HR=0.50), as well as PFS (8.1 vs 5.8 months; HR=0.42) respectively. The median OS with ACP (TECENTRIQ® along with Carboplatin and Paclitaxel) was 11.7 vs 9.9 months (HR=0.63), and PFS was 4.8 vs 5.8 months (HR=0.80), when compared with BCP (AVASTIN® plus Carboplatin and Paclitaxel). When compared to BCP, the ABCP group showed numerically greater survival than the ACP group among KRAS mutated patients. These results were consistent with reported survival improvements with immune checkpoint inhibitors in KRAS-mutant NSCLC.
In KRAS mutant patients across PD-L1 subgroups, OS and PFS were longer with ABCP when compared with BCP, but in PD-L1-low and PD-L1-negative subgroups, OS with ACP was similar to BCP. Conversely, in KRAS wild type patients, OS was longer with ACP than with ABCP or BCP across PD-L1 subgroups.
KRAS was frequently comutated with STK11, KEAP1, and TP53 and these subgroups conferred different prognostic outcomes. Within the KRAS mutated population, STK11 and/or KEAP1 mutations were associated with inferior OS and PFS across treatments compared with STK11-wild type and/or KEAP1wild type. In KRAS mutated patients with co-occurring STK11 and/or KEAP1 mutations (44.9%) or TP53 mutations (49.3%), survival was longer with ABCP than with ACP or BCP.
It was concluded that this analysis supported previous findings of mutation of STK11 and/or KEAP1 as poor prognostic indicators. Even though the clinical efficacy of ABCP (TECENTRIQ® and AVASTIN® along with Carboplatin and Paclitaxel) and ACP (TECENTRIQ® along with Carboplatin and Paclitaxel) was favorable compared with BCP (AVASTIN® plus Carboplatin and Paclitaxel) in these mutational subgroups, survival benefits were greater in the KRAS mutated and KEAP1 and STK11 wild type population versus KRAS mutated and KEAP1 and STK11 mutated population, suggesting both prognostic and predictive value of mutational analysis. The researchers added that these results suggest that TECENTRIQ® in combination with AVASTIN® and chemotherapy is an efficacious first-line treatment in metastatic NSCLC subgroups with KRAS mutations co-occurring with STK11 and/or KEAP1 or TP53 mutations and/or high PD-L1 expression.
Clinical efficacy of atezolizumab plus bevacizumab and chemotherapy in KRAS- mutated non-small cell lung cancer with STK11, KEAP1, or TP53 comutations: subgroup results from the phase III IMpower150 trial. West JH, McCleland M, Cappuzzo, F, et al. J Immunother Cancer. 2022 Feb;10(2):e003027. doi: 10.1136/jitc-2021-003027.
Segmentectomy versus Lobectomy in Small-Sized Peripheral Non-Small Cell Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers and Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Lobectomy is the standard of care for early-stage resectable Non-Small Cell Lung Cancer (NSCLC). Pneumonectomy is rarely performed due to unacceptably high mortality rate. Sublobar resection (Wedge resection or Segmentectomy) is considered a “compromise operation” in selected high risk patients with early stage lung cancer. With the approval of lung cancer screening in high risk individuals and subsequent detection of small tumors, Sublobar resections have been on the rise, even in good-risk patients in many institutions. Sublobar resection includes wedge resection and segmentectomy. In wedge resection, the lung tumor is removed with a surrounding margin of normal lung tissue, and is not an anatomical resection. Segmentectomy, unlike wedge resection, is an anatomical resection that usually includes one or more pulmonary parenchymal segments with the dissection of intraparenchymal and hilar lymph nodes. Wedge resection is inferior to anatomic segmentectomy and is associated with an increased risk of local recurrence and decreased survival in patients with Stage I NSCLC.
The clinical benefits and survival outcomes of segmentectomy have not been investigated in a randomized trial setting. The aim of this study was to investigate if segmentectomy was non-inferior to lobectomy in patients with small-sized peripheral NSCLC. In this randomized, controlled, multicenter, non-inferiority trial, 1106 patients (intention-to-treat population) were enrolled in Japan between Aug, 2009 and Oct 2014, and were randomly assigned 1:1 to receive either lobectomy (N=554) or segmentectomy (N=552). Enrolled patients had clinical Stage IA NSCLC based on contrast-enhanced CT scan and had a single tumor 2 cm or less in diameter, not located in the middle lobe, the center of which was in the outer third of the lung field, with no evidence of lymph node metastasis. Patient baseline clinicopathological factors were well balanced between the two treatment groups. The Primary endpoint was Overall Survival and Secondary endpoints included postoperative respiratory function at 6 months and 12 months, Relapse-Free Survival, proportion of local relapse and adverse events.
At a median follow up of 7.3 years, the 5-year Overall Survival was 94.3% for segmentectomy and 91.1% for lobectomy. Both superiority and non-inferiority in Overall Survival were confirmed using a stratified Cox regression model (HR=0.663; one-sided P<0.0001 for non-inferiority and P=0.0082 for superiority). This improved Overall Survival benefit was observed consistently across all predefined subgroups in the segmentectomy group. At 1 year follow-up, the significant difference in the reduction of median FEV1 between the two treatment groups was 3.5% (P<0.0001), but this however did not reach the predefined threshold for clinical significance of 10%. The 5-year Relapse-Free Survival was 88% for segmentectomy and 87.9% for lobectomy and was not statistically significant. The probability of local recurrence was approximately doubled and was 10.5% for segmentectomy and 5.4% for lobectomy (P=0.0018). Postoperative complications of grade 2 or worse occurred at similar frequencies in both treatment groups.
The authors concluded that this study is the first Phase III trial to show Overall Survival benefit with segmentectomy, compared to lobectomy, in patients with small-peripheral NSCLC. They added that segmentectomy should be the standard surgical procedure for this population of patients.
Segmentectomy versus lobectomy in small-sized peripheral non-small-cell lung cancer (JCOG0802/WJOG4607L): a multicentre, open-label, phase 3, randomised, controlled, non-inferiority trial. Saji H, Okada M, Tsuboi M, et al. The Lancet 2022;399:1607-1617.
Lung Cancer Screening with Low Dose CT Associated with Favorable Stage Shift and Improved Survival
SUMMARY: The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers and Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
In the National Lung Screening Trial (NLST) with Low Dose CT (LDCT) screening for lung cancer, there was a 20% reduction in mortality. Following the publication of the results of NLST, and NCCN issued guideline in 2011, the United States Preventive Services Task Force (USPSTF) recommended Lung Cancer screening with Low Dose CT scan in high risk patients. The CMS in 2015 determined that there was sufficient evidence to reimburse for this preventive service. The USPSTF expanded the criteria for Lung Cancer screening in 2021 and recommended annual screening with Low-Dose CT for adults aged 50 to 80 years who have a 20 pack-year smoking history and currently smoke or have quit within the past 15 years. The new USPSTF 2021 criteria were given a B recommendation, as there was additional research needed, to improve uptake of LDCT screening and to develop biomarkers to more accurately identify individuals, who would benefit from screening.
Approximately 15% of patients present with early stage (T1-2 N0) disease, and these numbers are likely to increase with the implementation of Lung Cancer screening programs. Surgical resection is the primary treatment for approximately 30% of patients with NSCLC who present with early Stage (I–IIIA) disease. In spite of the favorable stage shift as a result of lung cancer screening, low Health Care Provider knowledge of the lung cancer screening guidelines represents a potential barrier to implementation, and no clinical trials have shown these favorable benefits in a real world setting.
The authors in this study evaluated whether the introduction of Low Dose CT screening in 2013 resulted in an increase in the percentage of Stage I NSCLC diagnosed among patients potentially eligible for screening, along with an increase in median all cause survival among these patients, and whether any effects on stage extend to the entire study population or only select population groups. The researchers analyzed data from two large comprehensive US cancer registries-the National Cancer Database and the Surveillance Epidemiology End Results (SEER) program database using a quasi-experimental observational design. A total of 763 474 patients were identified for analysis in this study. They included those who were diagnosed as having NSCLC between 2010 and 2018 and who would have been eligible for screening by age criteria (age 55-79 years) and a comparator NSCLC patient cohort who would have been ineligible for screening (age 45-55). The authors then compared the rate of change in the percentage of patients with Stage I cancer at diagnosis between 2010 and 2018.
It was noted that among the screen eligible cohort of NSCLC patients, the percentage of patients with Stage I disease at diagnosis increased by 3.9% each year from 2014, following a minor change from 2010 to 2013. The rate of increase in Stage I diagnoses was more rapid in high lung cancer screening states. These findings however were not seen in the younger, screening ineligible patients. These results consistently noted across multiple analyses.
The median all cause survival of screening eligible patients aged 55-80 years increased at 11.9% per year from 2014 to 2018 (from 19.7 to 28.2 months). In multivariable adjusted analysis, the hazard of death decreased significantly faster after 2014 compared with before 2014 (P<0.001).
Disparities were however noted, and the benefits from this significant shift in the stage of the disease was not realized in racial or ethnic minority groups and those living in lower income or less educated regions. By 2018, Stage I NSCLC was the predominant diagnosis among non-Hispanic white people, whereas the economically deprived group of patients, were more likely to have Stage IV disease at diagnosis. Increases in the detection of early stage lung cancer in the US from 2014 to 2018 led to an estimated 10,100 averted deaths.
It was concluded from this study that although the adoption of lung cancer screening has been slow nationwide, this study indicated the beneficial effect of lung cancer screening and a recent stage shift toward Stage I NSCLC, with improved survival, following the introduction of lung cancer screening. This study also highlighted the disparities in the stage of lung cancer diagnosed between patient populations, reinforcing the need for equitable access to screening in the US.
Association of computed tomography screening with lung cancer stage shift and survival in the United States: quasi-experimental study. Potter AL, Rosenstein AL, Kiang MV, et al. BMJ 2022; 376 doi: https://doi.org/10.1136/bmj-2021-069008 (Published 30 March 2022)
Overall Survival at 2 Years with LUMAKRAS® for KRAS G12C Positive Non Small Cell Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The KRAS (kirsten rat sarcoma viral oncogene homologue) proto-oncogene encodes a protein that is a member of the small GTPase super family. The KRAS gene provides instructions for making the KRAS protein, which is a part of a signaling pathway known as the RAS/MAPK pathway. By relaying signals from outside the cell to the cell nucleus, the protein instructs the cell to grow, divide and differentiate. The KRAS protein is a GTPase, and converts GTP into GDP. To transmit signals, the KRAS protein must be turned on, by binding to a molecule of GTP. When GTP is converted to GDP, the KRAS protein is turned off or inactivated, and when the KRAS protein is bound to GDP, it does not relay signals to the cell nucleus. The KRAS gene is in the Ras family of oncogenes, which also includes two other genes, HRAS and NRAS. When mutated, oncogenes have the potential to change normal cells cancerous.
KRAS is the most frequently mutated oncogene in human cancers and are often associated with resistance to targeted therapies and poor outcomes. The KRAS-G12C mutation occurs in approximately 12-15% of Non Small Cell Lung Cancers (NSCLC) and in 3-5% of colorectal cancers and other solid cancers. KRAS G12C is one of the most prevalent driver mutations in NSCLC and accounts for a greater number of patients than those with ALK, ROS1, RET, and TRK 1/2/3 mutations combined. KRAS G12C cancers are genomically more heterogeneous and occur more frequently in current or former smokers, and are likely to be more complex genomically than EGFR mutant or ALK rearranged cancers. G12C is a single point mutation with a Glycine-to-Cysteine substitution at codon 12. This substitution favors the activated state of KRAS, resulting in a predominantly GTP-bound KRAS oncoprotein, amplifying signaling pathways that lead to oncogenesis.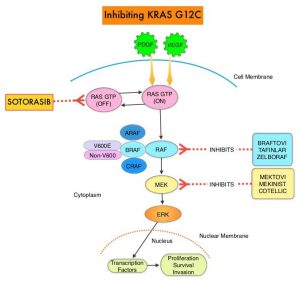
LUMAKRAS® (Sotorasib) is a first-in-class small molecule that specifically and irreversibly inhibits KRAS-G12C and traps KRAS-G12C in the inactive GDP-bound state. Preclinical studies in animal models showed that LUMAKRAS® inhibited nearly all detectable phosphorylation of Extracellular signal-Regulated Kinase (ERK), a key downstream effector of KRAS, leading to durable complete regression of KRAS-G12C tumors. The CodeBreaK clinical development program for LUMAKRAS® was designed to treat patients with an advanced solid tumor with the KRAS G12C mutation and address the longstanding unmet medical need for these cancers. This program has enrolled more than 800 patients across 13 tumor types since its inception.
CodeBreaK 100 is a Phase I and II, first-in-human, open-label, single arm, multicenter study, which enrolled patients with KRAS G12C-mutant solid tumors. Eligible patients must have received a prior line of systemic anticancer therapy, for their tumor type and stage of disease. The Phase II trial enrolled 126 patients with NSCLC, who had locally advanced or metastatic NSCLC with a KRAS G12C mutation, and had progressed on an immune checkpoint inhibitor and/or platinum-based chemotherapy. Patients with active brain metastases were excluded. Patient received LUMAKRAS® 960 mg orally once daily, until disease progression or unacceptable toxicity. The median age was 64 years, 52% were male, over 90% of patients had a smoking history, median number of prior lines of therapy was two, 92% had prior platinum-based chemotherapy and 90% had prior anti–PD-L1 therapy, 83% had both prior platinum-based chemotherapy and immunotherapy. The Primary end point of the trial was Overall Response Rate (ORR) as assessed by blinded Independent Central Review. Secondary end points included Duration of Response (DOR), Disease Control Rate (DCR), time to recovery, Progression Free Survival (PFS), Overall Survival (OS), and Safety. The examination of biomarkers served as an exploratory end point.
At the time of Primary analysis, at a median follow up of 12.2 months, the ORR was 37.1% and the median Duration of Response was 10 months. Based on the data from the primary analysis, the FDA in 2021 granted accelerated approval to LUMAKRAS®, for the treatment of patients with locally advanced or metastatic NSCLC, whose tumors harbor the KRAS G12C mutation, and who have received prior therapies.
For this updated analysis, the median follow up time for OS was 24.9 months, and the researchers included 174 patients enrolled in Phase I (N=48) and Phase II (N=126) portions of the CodeBreaK 100 trial, who were treated with LUMAKRAS®. The Overall Response Rate was 40.7% and the Disease Control Rate (DCR) was 83.7%. The median time to response was 6 weeks, the median Duration of Response was 12.3 month and 50.6% of responders remained in response for 12 months or more. The median PFS was 6.3 months and the median OS showed no change in the updated analysis, and was 12.5 months. At 1-year, the OS rate was 50.8% and the 2-year Overall Survival was 32.5%. The researchers performed additional analyses on both tumor and blood samples to identify biomarker profiles associated with durable clinical benefit and these showed that prolonged clinical benefit was observed regardless of Tumor Mutation Burden, PDL1 expression, and STK11 co-mutation status. Grade 3 or 4 treatment-related Adverse Events occurred in 21% of patients. Most adverse events were Grade 1 or 2, and treatment-related adverse events occurring in more than 10% of patients included diarrhea, elevated liver enzymes, nausea and fatigue.
It was concluded from this updated analysis that this is the longest follow up of patients on any KRAS G12C inhibitor, and LUMAKRAS® demonstrated meaningful and durable efficacy in patients with KRAS mutated NSCLC for whom treatment options are limited, following progression on first line treatment, and historically have had poor outcomes. Patients on LUMAKRAS® benefitted regardless of Tumor Mutation Burden, PDL1 expression, and STK11 co-mutation status. A global Phase III study (CodeBreaK 200) is underway, comparing LUMAKRAS® to Docetaxel in patients with KRAS G12C-mutated NSCLC.
Long-term outcomes with sotorasib in pretreated KRASp.G12C-mutated NSCLC: 2-year analysis of CodeBreaK100. Dy GK, Govindan R, Velcheti V, et al. Presented at: 2022 AACR Annual Meeting; April 8-13, 2022, New Orleans, LA. Abstract CT008.
FDA Approves Neoadjuvant OPDIVO® and Chemotherapy Combination for Early Stage Non Small Cell Lung Cancer
SUMMARY: The FDA on March 4, 2022, approved OPDIVO® (Nivolumab) with platinum-doublet chemotherapy for adult patients with resectable Non Small Cell Lung Cancer (NSCLC) in the neoadjuvant setting. This represents the first FDA approval for neoadjuvant therapy for early stage NSCLC. The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Surgical resection with a curative intent is the primary treatment for approximately 30% of patients with NSCLC who present with early Stage (I–IIIA) disease, unless medically unfit. These numbers are likely to increase with the implementation of Lung Cancer screening programs. These patients are often treated with platinum-based adjuvant chemotherapy/immunotherapy following surgical resection, to decrease the risk of recurrence. Nonetheless, 45-75% of these patients develop recurrent disease. There is therefore an unmet need for this patient population.
CHECKMATE-816 is an open-label, multicenter, randomized Phase III study which evaluated OPDIVO® plus chemotherapy versus chemotherapy alone as neoadjuvant treatment in patients with resectable Stage IB to IIIA NSCLC. In this trial, 358 patients with clinical Stage IB to Stage IIIA resectable NSCLC, with an ECOG Performance Status of 0 to 1 and no known sensitizing EGFR mutations or ALK alterations, were randomly assigned 1:1 to receive OPDIVO® at a dose of 360 mg IV along with platinum-doublet chemotherapy every 3 weeks for 3 doses (N=179) or chemotherapy alone on the same schedule (N=179). Patients then underwent radiologic staging and surgical resection within 6 weeks of neoadjuvant therapy. They then had the option of adjuvant therapy with or without radiation therapy, and were followed up. Both treatment groups were well balanced with regards to age, sex, histology and smoking status. About two-thirds of the patients had Stage IIIA disease. The median patient age was 65 years and patients were stratified by cancer stage, gender and PD-L1 status (1% or higher versus less than 1%). Tumor Mutational Burden results were available for 50% of patients. The Primary end points of this study were pathologic Complete Response (pCR), defined as the absence of viable tumor cells in lung and lymph nodes, and Event Free Survival (EFS). Secondary endpoints include major pathological response and Overall Survival. Key exploratory endpoints included feasibility of surgery and surgery-related adverse events.
The pCR rate was 24% in the OPDIVO® plus chemotherapy group and 2.2% in the chemotherapy alone group. The pCR improvement was noted with the OPDIVO® plus chemotherapy combination regardless of disease stage and irrespective of radiologic downstaging. Overall, 83% of patients assigned to OPDIVO® plus chemotherapy and 78% of patient’s assigned to chemotherapy alone achieved R0 resection, with 10% versus 74% median residual viable tumor cells noted in the primary tumor bed respectively. Lung-sparing surgery (lobectomy) was performed in 77% of patients assigned to OPDIVO® plus chemotherapy versus 61% among those assigned to chemotherapy alone. The median EFS was 31.6 months in the OPDIVO® plus chemotherapy group and 20.8 months for those receiving chemotherapy alone (HR=0.63; P=0.0052).
The authors concluded that CheckMate 816 is the first Phase III trial to show a benefit for neoadjuvant immunotherapy plus platinum-doublet chemotherapy in earlier stage resectable NSCLC, with marked improvement in pathologic Complete Response rate, without any meaningful increase in toxicity or decrease in the feasibility of surgery. It is likely that the higher pathologic Complete Response rate may translate into higher cure rates, with longer follow up.
Surgical outcomes from the phase 3 CheckMate 816 trial: nivolumab (NIVO) + platinum-doublet chemotherapy (chemo) vs chemo alone as neoadjuvant treatment for patients with resectable non-small cell lung cancer (NSCLC). Spicer J, Wang C, Tanaka F, et al. J Clin Oncol. 2021;39(suppl 15):8503. doi:10.1200/JCO.2021.39.15_suppl.8503
Smoking Cessation after Lung Cancer Diagnosis Improves Overall Survival
SUMMARY: According to the American Cancer Society, tobacco use is responsible for about 1 in 5 deaths in the United States and is the leading preventable cause of death in the US. Smoking (cigarettes, cigars, and pipes) is responsible for about 20% of all cancers and about 30% of all cancer deaths in the US. Approximately 80% of lung cancers, as well as about 80% of all lung cancer deaths, are due to smoking, and lung cancer is the leading cause of cancer death in both men and women. Smoking also increases the risk for cancers of the Oral cavity, Oropharynx, Larynx, Esophagus, Stomach, Liver, Pancreas, Colon/Rectum, Kidney, Bladder, Cervix, as well as Acute Myeloid Leukemia. The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Previous published studies have shown that individuals who start smoking at a younger age have greater mortality risk than those who start smoking later in life, and smoking cessation especially at younger ages substantially reduces that mortality risk. Several biologic mechanisms have been hypothesized, to explain the beneficial effect of smoking cessation on survival, in patients with Lung Cancer. Tobacco smoke has been shown to promote tumor growth, progression, and dissemination, while decreasing the efficacy and tolerance to radiation and systemic therapy. Further, it is well established that smoking increases the risk of postoperative complications and second primary cancers. Epigenetic changes induced by tobacco smoke may play an important role, and cigarette smoke induced diseases may be the result of alterations in DNA methylation, and is a reversible gene regulatory modification. Following smoking cessation, the majority of the differentially methylated CpG dinucleotide sites of the current smokers return to the level of the never smokers within 5 years of smoking cessation. However, some of the methylated genes may not return to the level of the never smokers even after 30 years of smoking cessation, suggesting that tobacco smoke can lead to lasting damage to human health. In this publication, the researchers aimed to summarize the current scientific evidence on whether quitting smoking at or around diagnosis has a beneficial effect on the survival of Lung Cancer patients.
The authors conducted a systematic literature review and meta-analysis of the studies that evaluated the prognostic effect of quitting smoking at or around diagnosis among patients with Lung Cancer. The meta-analysis included 21 articles published between 1980 and October 2021 on the effect of smoking cessation at or around the time of diagnosis among a total of 10,938 patients with lung cancer, which included patients with Non Small Cell Lung Cancer, Small Cell Lung Cancer, as well as patients with Lung Cancer of both or unspecified subtypes or whose subtype was not specified. In most studies analyzed, the median age at Lung Cancer diagnosis was between 60 and 70 years. The authors used random effect meta-analysis models to pool study-specific data into Summary Relative Risk (SRR) and corresponding 95% confidence intervals (CI). There was substantial variability across studies with regards to study design, patient characteristics, treatments received, criteria used to define smoking status (quitters or continued), and duration of follow up.
Even though there was moderate heterogeneity of Hazard Ratio across studies, it was noted that quitting smoking at or around diagnosis was associated with a significant 29% improvement in Overall Survival, compared with patients who continued to smoke after their diagnosis (SRR=0.71; 95% CI 0.64–0.80). This benefit of quitting smoking was noted regardless of lung cancer histologic subtype, with a 20-30% reduction in the risk of death among those who quit smoking post-diagnosis, compared to those who continued to smoke.
It was concluded from this analysis that quitting smoking at or around diagnosis is associated with a beneficial effect on the survival of Lung Cancer patients, and smoking cessation can be nearly as effective in improving the chance of survival as treatment with chemotherapy, immunotherapy or radiation therapy. The authors added that based on these finding, Health Care Providers should educate Lung Cancer patients about the benefits of quitting smoking even after diagnosis and provide them with the necessary support for smoking cessation.
Quitting Smoking At or Around Diagnosis Improves the Overall Survival of Lung Cancer Patients: A Systematic Review and Meta-Analysis. Caini S, Riccio MD, Vettori V, et al. Published:January 04, 2022. DOI:https://doi.org/10.1016/j.jtho.2021.12.005
NUBEQA® Combination Improves Overall Survival in Metastatic Hormone Sensitive Prostate Cancer
SUMMARY: Prostate cancer is the most common cancer in American men with the exclusion of skin cancer, and 1 in 9 men will be diagnosed with Prostate cancer during their lifetime. It is estimated that in the United States, about 268,490 new cases of Prostate cancer will be diagnosed in 2022 and 34,500 men will die of the disease. The development and progression of Prostate cancer is driven by androgens. Androgen Deprivation Therapy (ADT) or testosterone suppression has therefore been the cornerstone of treatment of advanced Prostate cancer and is the first treatment intervention.
The first-generation NonSteroidal Anti-Androgen (NSAA) agents such as EULEXIN® (Flutamide), CASODEX® (Bicalutamide) and NILANDRON® (Nilutamide) act by binding to the Androgen Receptor (AR) and prevent the activation of the AR and subsequent up-regulation of androgen responsive genes. They may also accelerate the degradation of the AR. These agents have a range of pharmacologic activity from being pure anti-androgens to androgen agonists. CASODEX® is often prescribed along with GnRH (Gonadotropin-Releasing Hormone) agonists for metastatic disease, or as a single agent second line hormonal therapy for those who had progressed on LHRH agonists.
NUBEQA® (Darolutamide) is a potent second-generation Androgen Receptor (AR) inhibitor with a new chemical structure and has a high affinity to the AR. NUBEQA® does not cross the blood-brain barrier and for this reason has a favorable safety and tolerability profile in prespecified adverse events such as seizures, when compared with other second-generation AR inhibitors such as ERLEADA® (Apalutamide) and XTANDI® (Enzalutamide). It has been associated with increased Overall Survival (OS) among patients with non-metastatic Castration-Resistant Prostate Cancer (CRPC) and has been approved by the FDA for this indication. Whether a combination of NUBEQA®, in combination with Androgen Deprivation Therapy (ADT), and Docetaxel would increase survival among patients with metastatic Hormone-Sensitive Prostate Cancer, is unknown.
ARASENS is an international, randomized, double-blind, placebo-controlled, Phase III trial, which evaluated the efficacy and safety of NUBEQA® (Darolutamide) added to Androgen Deprivation Therapy (ADT) and Docetaxel in patients with metastatic Hormone Sensitive Prostate Cancer. In this study, a total of 1306 patients were randomly assigned 1:1 to receive NUBEQA® (N=651) or placebo (N=655), both in combination with ADT and Docetaxel. All the patients received ADT (either a Luteinizing Hormone Releasing Hormone (LHRH} agonist or antagonist) or underwent Orchiectomy within 12 weeks before randomization and received six cycles of Docetaxel 75 mg/m2 IV given on Day 1 every 21 days, with Prednisone or Prednisolone. Patients received LHRH agonists along with a first-generation anti-androgen agent for at least 4 weeks before randomization to help prevent a tumor flare, and the anti-androgen agent was discontinued before randomization. Patients were then randomly assigned to receive either NUBEQA® 600 mg orally twice daily or matched placebo, and treatment was continued until disease progression or unacceptable toxicities.
Eligible patients had biopsy proven prostate cancer with bone metastases and had to be candidates for ADT and Docetaxel. Patients with regional lymph node involvement only (N1, below the aortic bifurcation) or if they had received ADT more than 12 weeks before randomization, second-generation Androgen Receptor pathway inhibitors, chemotherapy, or immunotherapy for prostate cancer before randomization, or radiotherapy within 2 weeks before randomization, were excluded. The median age was 67 years and both treatment groups were well balanced. All patients had metastatic disease at baseline, 78% of the patients had a Gleason score of 8 or greater, about 80% had bone metastases (Stage M1b) and 18% had visceral metastases (Stage M1c). The Primary end point was Overall Survival (OS) and Secondary end points included were time to Castration-Resistant Prostate Cancer, time to pain progression, symptomatic Skeletal Event-Free Survival and time to initiation of subsequent systemic antineoplastic therapy, as well as Safety. The median follow up for Overall Survival was 43 months.
The median Overall Survival was not estimable in the NUBEQA® group versus 48.9 months in the placebo group. The addition of NUBEQA® to the combination with ADT and Docetaxel reduced the risk of death by 32%, compared to the placebo group (HR=0.68; P<0.001). This OS benefit was noted across most subgroups. Further, the significant OS benefit with the addition of NUBEQA® was observed, despite receipt of subsequent life-prolonging systemic therapies such as different Androgen-Receptor pathway inhibitors by 75.6% of patients in the placebo control group. The OS at 4 years was 62.7% in the NUBEQA® group and 50.4% in the placebo group.
With regard to Secondary endpoints, the addition of NUBEQA® to ADT and Docetaxel demonstrated consistent benefits. The time to development of Castration-Resistant Prostate Cancer was significantly longer in the NUBEQA® group (HR=0.36; P<0.001), the time to pain progression was also significantly longer in the NUBEQA® group (HR=0.79; P=0.01), as well as symptomatic Skeletal Event-Free Survival (HR=0.61; P<0.001). Further, the time to the initiation of subsequent systemic antineoplastic therapy was also significantly longer in the NUBEQA® group (HR=0.39; P<0.001). Adverse events were similar in the two groups.
The authors concluded that among patients with metastatic Hormone Sensitive Prostate Cancer, the addition of NUBEQA® to Androgen Deprivation Therapy and Docetaxel resulted in significantly longer Overall Survival, as well as improvement in key Secondary end points, with no increase in adverse events.
Darolutamide and Survival in Metastatic, Hormone-Sensitive Prostate Cancer. Smith MR, Hussain Saad F, et al. for the ARASENS Trial Investigators. NEJM. February 17, 2022. DOI: 10.1056/NEJMoa2119115.
ENHERTU® in HER2-Mutant Non Small Cell Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. HER2 is a Tyrosine Kinase Receptor expressed on the surface of several tumor types including Breast, Gastric, Lung and Colorectal cancers. It is a growth-promoting protein and HER2 overexpression/HER2 gene amplification is often associated with aggressive disease and poor prognosis in certain tumor types. However, HER2 overexpression and gene amplification are associated with distinct molecular entities and have limited therapeutic value in lung cancer.
HER2 mutations unlike HER2 overexpression and gene amplification are oncogenic drivers, and have been detected in 2 to 3% of NSCLCs. They are more often detected in female patients and never-smokers, and almost exclusively in Adenocarcinomas. Majority of of HER2 mutations (80-90%) occur in exon 20, as either a duplication or an insertion of 12 nucleotides, resulting in the addition of four amino acids (YVMA) at codon 775 in the kinase domain. This distinct molecular entity is characterized by specific pathological and clinical behavior. These acquired HER2 gene mutations have been independently associated with cancer cell growth and poor prognosis. There are currently no therapies approved specifically for the treatment HER2 mutant NSCLC, and is therefore an unmet need.
ENHERTU® (Trastuzumab Deruxtecan) is an Antibody-Drug Conjugate (ADC) composed of a humanized monoclonal antibody specifically targeting HER2, with the amino acid sequence similar to HERCEPTIN® (Trastuzumab), attached to a potent cytotoxic Topoisomerase I inhibitor payload by a cleavable tetrapeptide-based linker. ENHERTU® has a favorable pharmacokinetic profile and the tetrapeptide-based linker is stable in the plasma and is selectively cleaved by cathepsins that are up-regulated in tumor cells. Unlike KADCYLA® (ado-Trastuzumab emtansine), which is also an Antibody-Drug Conjugate, ENHERTU® has a higher drug-to-antibody ratio (8 versus 4), the released payload easily crosses the cell membrane with resulting potent cytotoxic effect on neighboring tumor cells regardless of target expression, and the released cytotoxic agent (payload) has a short half-life, minimizing systemic exposure. ENHERTU® is approved in the US for the treatment of adult patients with unresectable or metastatic HER2 positive breast cancer who received two or more prior anti-HER2 based regimens, and locally advanced or metastatic HER2-positive Gastric or GastroEsophageal Junction adenocarcinoma who have received a prior Trastuzumab based regimen. Translational research demonstrated that HER2-mutant NSCLC may preferentially internalize the HER2 receptor Antibody-Drug Conjugate complex regardless of HER2 protein expression, and overcome resistance to other HER2-targeted agents.
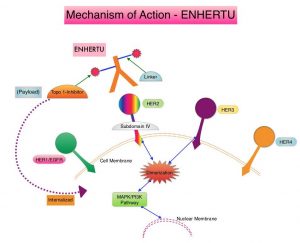
DESTINY-Lung01 is a global, multicenter, open-label, two-cohort, Phase II study, conducted to evaluate the efficacy and safety of ENHERTU® in patients with HER2 mutant or HER2 overexpressing (defined as ImmunoHistoChemistry-IHC 3+ or IHC 2+), unresectable and metastatic non-squamous NSCLC. A total of 91 patients with HER2-mutant NSCLC were enrolled between May 30, 2018, and July 21, 2020 and treated with ENHERTU®. Patients who had previously been treated with a HER2 antibody or an Antibody-Drug Conjugate were ineligible for participation, but those who had previously received a HER2 Tyrosine Kinase Inhibitor such as Afatinib, Pyrotinib, or Poziotinib were eligible. The median patient age was 60 yrs and enrolled patients had a median of two prior lines of therapy, with majority of patients having received platinum-based chemotherapy (95%) and anti-PD-1 or PD-L1 treatment (66%). About 20% of patients received Docetaxel and 14% received HER TKIs. For the majority of patients (93%), HER2 mutations were located in the kinase domain. Patients received ENHERTU® 6.4 mg/kg every 3 weeks by intravenous infusion. The Primary endpoint was Objective Response Rate (ORR) as assessed by Independent Central Review. Secondary endpoints included Disease Control Rate (DCR), Duration of Response (DoR), Progression Free Survival (PFS), Overall Survival (OS) and Safety. At the time of data cutoff, the median duration of treatment was 6.9 months and treatment was ongoing for 16% of patients.
At a median follow up 13.1 months, the ORR was 55% and the median Duration of Response was 9.3 months. Responses were observed across different HER2 mutation subtypes. The median PFS was 8.2 months and the median OS was 17.8 months. The most common Grade 3 or higher drug-related Adverse Event was neutropenia noted in 19% of patients and adjudicated drug-related Interstitial Lung Disease occurred in 26% of patients and resulted in 2 deaths.
It was concluded that ENHERTU® demonstrated promising clinical activity, with a high Objective Response Rate and durable responses, in a heavily pretreated population of patients with HER2-mutated NSCLC.
Trastuzumab Deruxtecan in HER2-Mutant Non–Small-Cell Lung Cancer. Li BT, Smit EF, Goto Y, et al., for the DESTINY-Lung01 Trial Investigators. N Engl J Med 2022; 386:241-251
Blood Test Plus Risk Model May Predict Who May Benefit From Lung Cancer Screening
SUMMARY: The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers and Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
In the National Lung Screening Trial (NLST) with Low Dose CT (LDCT) screening for lung cancer, there was a 20% reduction in mortality. Following the publication of the results of NLST, and NCCN issued guideline in 2011, the United States Preventive Services Task Force (USPSTF) recommended Lung Cancer screening with Low Dose CT scan in high risk patients. CMS in 2015 determined that there was sufficient evidence to reimburse for this preventive service. The USPSTF expanded the criteria for Lung Cancer screening in 2021 and recommended annual screening with Low-Dose CT for adults aged 50 to 80 years who have a 20 pack-year smoking history and currently smoke or have quit within the past 15 years. The new USPSTF 2021 criteria were given a B recommendation, as there was additional research needed, to improve uptake of LDCT screening and to develop biomarkers to more accurately identify individuals, who would benefit from screening.
The authors in a previously published study identified the precursor form of Surfactant Protein B (Pro-SFTPB) as predictive of Lung Cancer risk. They were also able to, in a proof-of-principle study, demonstrate that a four Marker Protein panel (4MP) consisting of Pro-SFTPB, Cancer Antigen 125, CarcinoEmbryonic Antigen, and Cytokeratin-19 fragment, has the potential to identify individuals at risk for developing Lung Cancer, better than Pro-SFTPB alone.
The purpose of this study was to determine whether a blood-based four Marker Protein biomarker panel together with the PLCOm2012 Lung Cancer prediction model would better identify individuals for Lung Cancer screening, compared with current US Preventive Services Task Force (USPSTF) criteria.
The PLCO Cancer Screening Trial was a randomized multicenter trial which evaluated the impact of screening for Prostate, Lung, Colorectal, and Ovarian cancer, on disease-specific mortality. In this study, a biorepository was created for blood specimens that were annually collected, after obtaining consent from the intervention group participants. In this study there were 42,450 individuals in the intervention group who have ever smoked, and 85% of the participants in this intervention arm had at least one blood specimen collection. Individuals with histologically-confirmed lung cancers from the ever-smoked participants in the intervention group, who were diagnosed within 6 years of study entry, and with pre-diagnostic blood specimen available (N= 552), were included in the current study.
Non cancer participants who have ever smoked (N=2193) were randomized 4:1 with lung cancer diagnosed cases and were followed for an additional 13 years during which time they remained cancer-free. For each selected participant, all blood samples collected within 6 years of study entry, or up to the time of diagnosis for lung cancer cases, were included in the study specimen set (N=10,008 blood specimens). The mean age of the study population was 65 years, and 64% were male, 60% were former smokers and 40% were current smokers. The baseline information for the PLCOm2012 model included age, race or ethnic group, education, Body Mass Index, Chronic Obstructive Pulmonary Disease, personal history of cancer, family history of lung cancer and smoking status (current versus former), intensity, duration, and quit time. Levels of the 4 marker proteins in the serum were determined using bead-based immunoassays and biomarker scores were calculated for the combined 4 marker proteins. The researchers assessed the performance of the 4 marker protein panel in combination with the PLCOm2012 lung cancer prediction model, and compared the combination with current USPSTF lung cancer screening criteria, among pre-diagnosis lung cancer, and non-lung cancer serum samples, from the PLCO Cancer Screening Trial.
Using prediagnostic case, and non-case serum samples from the PLCO Cancer Screening Trial data, a combined four-marker protein panel along with PLCO m2012 model showed statistically significant improvement in sensitivity by 11.9% and 9.9% and specificity by 12.9% and 6.9% ,compared with USPSTF 2013 and the recent USPSTF 2021 criteria, respectively. If the 4MP along with PLCOm2012 model was applied to individuals with 10 or more pack-year smoking history, the improved performance of this combination would have resulted in referral to screening of 12.6% more lung cancer cases among the 119 cases who would otherwise receive a lung cancer diagnosis within a year, as well as nonreferral of 29.6% of the 14,061 non-cases. When compared with the USPSTF 2021 criteria, the 4MP plus PLCOm2012 model would have identified for annual screening 9.2% more lung cancer cases and would have reduced referral by 13.7% among non-cases, compared with USPSTF 2021 criteria.
It was concluded that the 4MP plus PLCOm2012 model yielded superior predictive performance and sensitivity and specificity for ruling individuals into LDCT screening, compared with USPSTF 2013 or USPSTF 2021 eligibility criteria, and with the PLCOm2012 model alone. The authors added that the public health benefit is significant, as it better identifies individuals at high risk of lung cancer and expands upon the number of individuals who would be considered eligible for lung cancer screening, thereby addressing some of the limitations of current screening eligibility criteria. These findings have important implications for improving lung cancer screening programs and reducing the burden of lung cancer through personalized risk assessment.
Blood-Based Biomarker Panel for Personalized Lung Cancer Risk Assessment. Fahrmann JF, Marsh T, Irajizad E, et al. J Clin Oncol. Published online January 7, 2022. doi:10.1200/JCO.21.01460.
Adherence to Guideline-Recommended Biomarker Testing as an Integral Component of NSCLC Care
Written by: Dr. David M. Waterhouse, M.D., MPH
Content Sponsored by: Amgen
The NSCLC Landscape Has Evolved Significantly Due Largely to the Growing Number of Actionable Mutations1
Despite innovations in standard-of-care, advanced non-small cell lung cancer (NSCLC) continues to burden patients, with poor survival outcomes.2,3 NSCLC has been identified as the leading cause of cancer death worldwide with an estimated 1.8 million deaths in 2020.2 As the number of targeted therapies and approval companion diagnostics continues to grow, mortality and survival rates have begun to improve.3 With the addition of KRAS G12C, there are 9 actionable molecular biomarkers (as of October 2021) and more than 20 targeted therapies approved for use in advanced NSCLC.1,4 Guidelines recommend biomarker testing for all eligible patients at diagnosis of advanced NSCLC regardless of characteristics such as smoking history, race, or sex.5,6 Unfortunately, real-world evidence shows that far too many patients fail to receive comprehensive biomarker testing, despite their eligibility.7,8
Adherence to Guidelines Can Improve Biomarker Testing9
As targeted therapies are approved, guidelines continue to update their recommendations on biomarker testing.5 As of December 2021, NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC recommend broad molecular testing of actionable and emerging biomarkers for patients with advanced or metastatic NSCLC (Figure 1).5 Similarly, the American Society of Clinical Oncology (ASCO) endorsed the 2018 College of American Pathologists (CAP)/International Association for the Study of Lung Cancer (IASLC)/Association for Molecular Pathology (AMP) guidelines, recommending comprehensive cancer panel testing for genetic biomarkers.10,11
Figure 1: NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC5,*,†
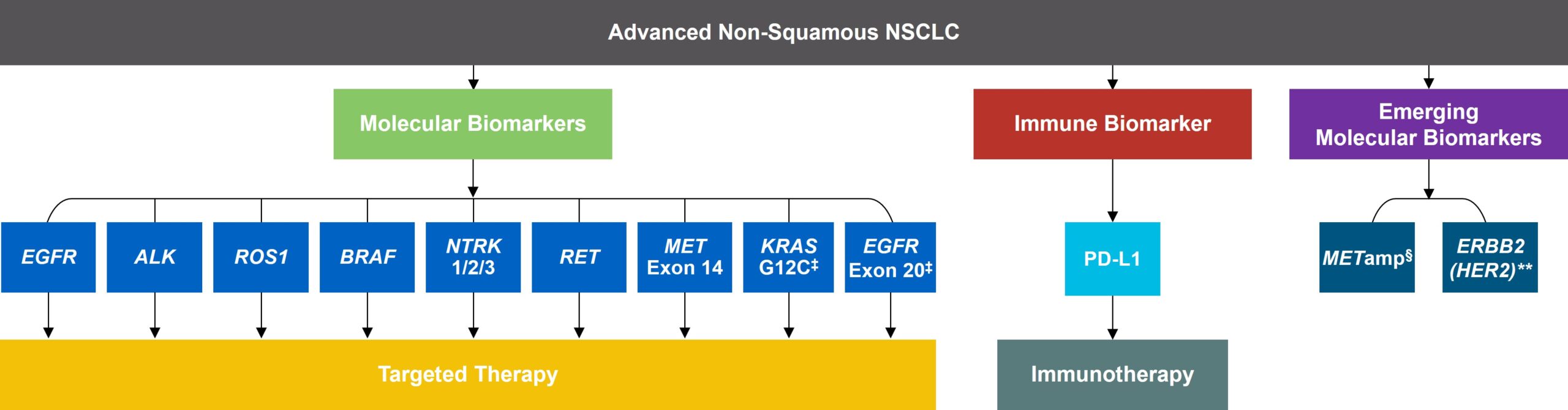 *The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC provide recommendations for certain individual biomarkers that should be tested and recommend testing techniques but do not endorse any specific commercially available biomarker assays or commercial laboratories.5
*The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC provide recommendations for certain individual biomarkers that should be tested and recommend testing techniques but do not endorse any specific commercially available biomarker assays or commercial laboratories.5
†The NCCN Guidelines® for NSCLC recommend broad molecular testing to identify rare driver variants for which targeted therapies may be available to ensure patients receive the most appropriate treatment.5
‡KRAS G12C and EGFR exon 20 mutations are used to determine subsequent (ie, second-line and beyond) therapy using targeted agents or other novel agents.5
§The definition of high-level MET amplification is evolving and may differ according to the assay used for testing. For NGS-based results, a copy number greater than 10 is consistent with high-level MET amplification.5
**For oncogenic or likely oncogenic HER2 mutations, refer to definitions at oncokb.org.5
Although adherence to guideline-recommended biomarker testing is associated with improved patient outcomes, real-world EMR data reveals suboptimal biomarker testing rates.7,9 In a retrospective study, 81% of patients with metastatic NSCLC did not receive testing for ALK, EGFR, ROS1, and BRAF before initiation of first-line treatment, despite the availability of targeted therapies.7 Moreover, only 28% of patients received testing for all four genetic biomarkers and PD-L1 during the study period.7 In another retrospective study, less than 50% of patients with metastatic NSCLC received testing for all five biomarkers (EGFR, ALK, ROS1, BRAF, PD-L1) (Figure 2).8
Beyond the underutilization of biomarker testing, there remains an even greater need to increase broad molecular testing among racial and ethnic minority groups in the US.12,13 In one retrospective study, Black/African American patients with advanced NSCLC had significantly lower rates of testing with NGS assays (43.8%) compared with White patients (54.7%) (Figure 3).12
Figure 2: MYLUNG Consortium™ EMR Analysis of Patients With Metastatic NSCLC8,††
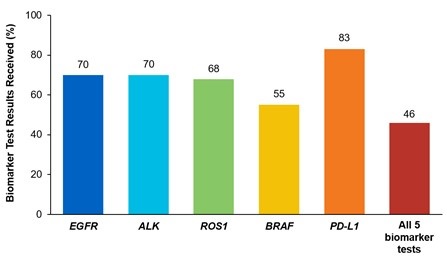
††A retrospective, observational study assessing real-world biomarker testing patterns in patients with metastatic NSCLC from community oncology practices within the US Oncology Network community practices between 2018 and 2020.8
Figure 3: EMR Analysis of Biomarker Testing in Patients With Advanced/Metastatic NSCLC12,‡‡
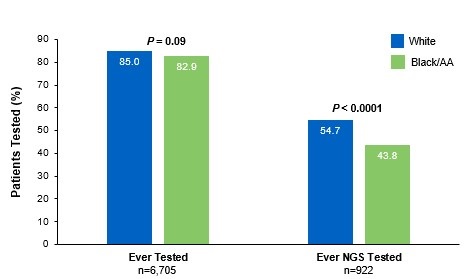
‡‡A retrospective cohort study of patients with advanced/metastatic non-squamous NSCLC (N=10,333) from ~ 800 sites of care in the US identified via the Flatiron Electronic Health Record Database between 2017 and 2020.12
Collectively, these findings highlight the disparity in proactive disease management across different patient populations.7,8,12
Considerations Across the Biomarker Testing Journey
There are several different methods in which eligible patients can be tested for actionable genetic alterations, each with unique considerations as indicated below (Figure 4).
Figure 4: Comparing Biomarker Testing Methods and Sample Types1,6,11,14,15
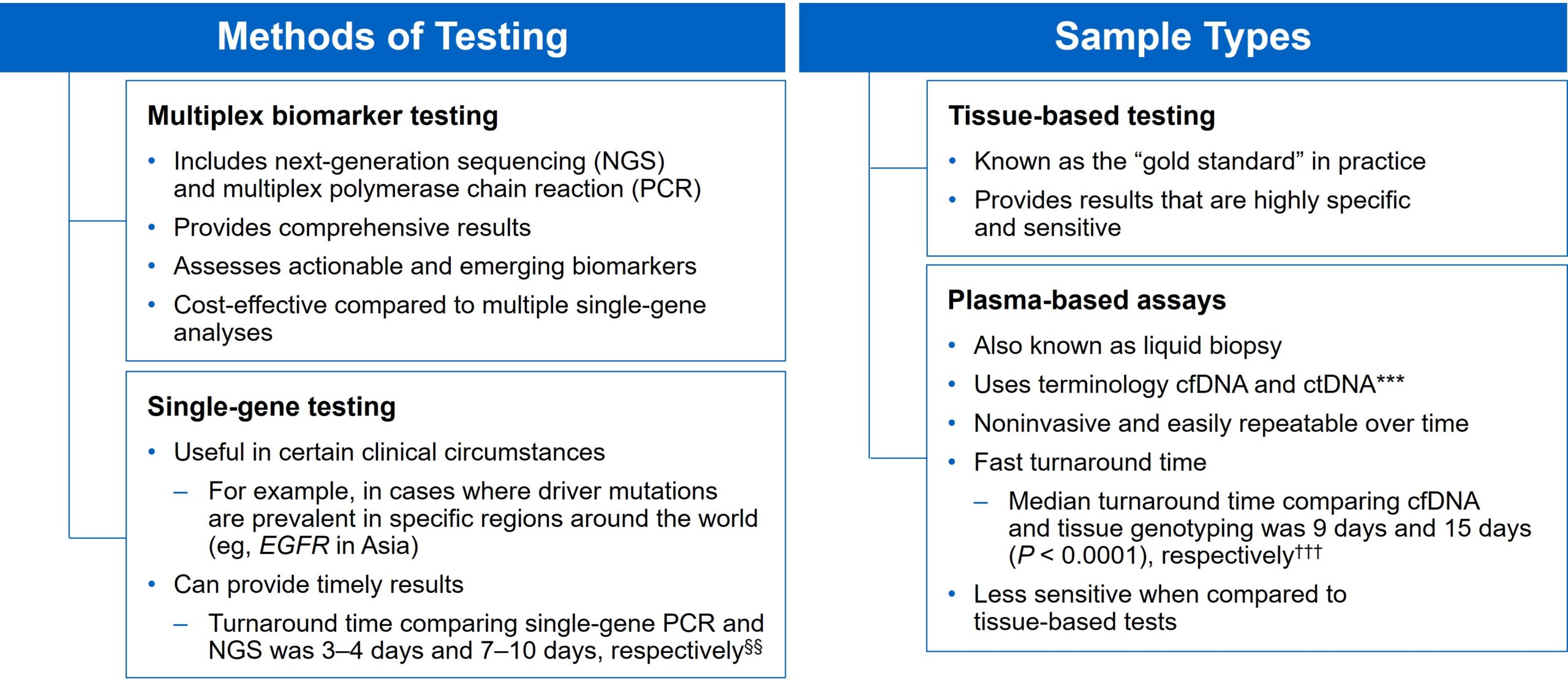
§§Data from a review of common molecular assays for biomarker testing that analyzed common detected variants, sensitivities, and turnaround time.6
***cfDNA refers to all circulating DNA (largely non-malignant), while ctDNA refers to the tumor-related component of cfDNA.14
†††Data from a prospective study that enrolled patients with previously untreated metastatic NSCLC undergoing SOC tissue genotyping and comprehensive cfDNA analysis, with turnaround time defined as the number of days between the test order date and the retrieval of test results.15
While tissue biopsy remains the “gold standard” in NSCLC, it may not be feasible (insufficient tissue) or pragmatic (urgent need to begin treatment) in all patients.6,14,16,17 Plasma ctDNA demonstrates complementary results to tissue-based assays and can be considered a valid tool for genotyping of newly diagnosed patients with advanced NSCLC.14 In a prospective study from 2016 to 2018, guideline-recommended biomarkers with FDA-approved therapies (EGFR Exon 19 deletion and L858R, ALK fusion, ROS1 fusion, BRAF V600E) showed ≥ 98.2% concordance between tissue and liquid-based testing.15 While concordance is high for any single test, high concordance for full panels will be required for liquid biopsies to become standard. Additionally, negative results on liquid biopsy still require validation with tissue testing.14,15,17
Liquid biopsy offers improvements in sample acquisition and small tissue samples and provides less invasive procedures and shortened turnaround times.14,15,18 Other considerations for maximizing the tissue journey include the use of comprehensive testing, rapid on-site evaluation (ROSE), and implementing reflex testing protocols with the help of a multidisciplinary team (MDT).6,17,18
Delays in Biomarker Testing Results May Impact Treatment Decisions16
Longer turnaround times for molecular testing compared with turnaround times for PD-L1 testing by IHC may result in the initiation of immunotherapy before molecular testing results are received.16 Waiting for complete biomarker test results prior to initiating therapy can allow doctors to make the most informed decisions surrounding a patient’s treatment journey.16 The benefits of delaying therapy may outweigh the risks associated with initiating immunotherapy treatment in patients with actionable genetic alterations who are eligible for targeted treatments, such as immunotherapy treatments, which are associated with a delay in benefit when compared to targeted therapy.16
Ensuring Best Practices in Biomarker Testing Can Lead to Better Patient Outcomes9
As the NSCLC landscape continues to progress with the increasing number of actionable biomarkers, there is a growing need for proactive and comprehensive molecular testing.1,7,8,12,17 Although real-world data has shown significant underuse of biomarker testing, rates can be improved with a diligent observation of expanding guidelines and recommendations by expert panels and associations.7-9,12 In the coming years, it will be increasingly urgent for clinicians to evolve institutional protocols, including enabling reflex testing, and work as an MDT to ensure biomarker testing is performed on all eligible patients with advanced NSCLC.6,17,18
[Abbreviations]
ALK, anaplastic lymphoma kinase; BRAF, proto-oncogene B-Raf; cfDNA, cell-free DNA;
ctDNA, circulating tumor DNA; EGFR, epidermal growth factor receptor; EMR, electronic medical record; ERBB2, erb-b2 receptor tyrosine kinase 2; HER2, human epidermal growth factor receptor 2;
IHC, immunohistochemistry; KRAS, Kirsten rat sarcoma viral oncogene homolog; MET, mesenchymal-to-epithelial transition; mNSCLC, metastatic non-small cell lung cancer; NCCN, National Comprehensive Cancer Network; NTRK, neurotrophic tyrosine receptor kinase; PD-L1, programmed cell death ligand 1; RET, rearranged during transfection; ROS1, c-ros oncogene 1; SOC, standard-of-care.
[References]
1. Majeed U, et al. J Hematol Oncol. 2021;14:108.
2. Sung H, et al. CA Cancer J Clin. 2021;71:209-249.
3. Siegel RL, et al. CA Cancer J Clin. 2021;71:7-33.
4. Food and Drug Administration. www.fda.gov. Accessed October 6, 2021.
5. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer. v.1.2022. ©National Comprehensive Cancer Network, Inc. 2021. All rights reserved. Accessed December 14, 2021. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
6. Pennell NA, et al. Am Soc Clin Oncol Educ Book. 2019;39:531-542.
7. Nadler ES, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 9079.
8. Robert NJ, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 102.
9. John A, et al. Adv Ther. 2021;38:1552-1566.
10. Hanna N, et al. J Clin Oncol. 2017;35:3484-3515.
11. Lindeman NI, et al. Arch Pathol Lab Med. 2018;142:321-346.
12. Bruno DS, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 9005.
13. Hann KEJ, et al. BMC Public Health. 2017;17:503.
14. Rolfo C, et al. J Thorac Oncol. 2021;16:1647-1662.
15. Leighl NB, et al. Clin Cancer Res. 2019;25:4691-4700.
16. Smeltzer MP, et al. J Thorac Oncol. 2020;15:1434-1448.
17. Gregg JP, et al. Transl Lung Cancer Res. 2019;8:286-301.
18. VanderLaan PA, et al. Cancer Cytopathol. 2020;128:629-636.
USA-510-80864 12/21
KRAS Variant Status and Outcomes with Immune Checkpoint Inhibitor-Based Therapy in Advanced Non Small Cell Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Patients with advanced NSCLC without a driver mutation and with Programmed cell Death Ligand 1 (PD-L1) expression of 50% or greater, are often treated first line with Immune Checkpoint Inhibition (ICI) monotherapy or ICI in combination with chemotherapy. The choice between these two treatment regimens is usually based on tumor burden and patient comorbidities, as there are presently no biomarkers available to predict the risk and benefit of these treatment interventions. The KEYNOTE-042 study demonstrated that single agent Pembrolizumab given as first line therapy demonstrated Overall Survival (OS) benefit over chemotherapy, in patients with previously untreated advanced NSCLC, with PD-L1 expression of 1% or greater. In an exploratory analysis, this benefit was seen regardless of KRAS status, but was more pronounced in patients with KRAS variants than those without KRAS variants.
The KRAS (kirsten rat sarcoma viral oncogene homologue) proto-oncogene encodes a protein that is a member of the small GTPase super family. The KRAS gene provides instructions for making the KRAS protein, which is a part of a signaling pathway known as the RAS/MAPK pathway. When mutated, KRAS oncogene has the potential to change normal cells cancerous. KRAS is the most frequently mutated oncogene in human cancers and are often associated with resistance to targeted therapies and poor outcomes. The KRAS-G12C mutation occurs in approximately 12-15% of NSCLC and in 3-5% of Colorectal cancers and other solid cancers. KRAS G12C is one of the most prevalent driver mutations in NSCLC and accounts for a greater number of patients than those with ALK, ROS1, RET, and TRK 1/2/3 mutations combined. KRAS G12C cancers are genomically more heterogeneous and occur more frequently in current or former smokers, and are likely to be more complex genomically than EGFR mutant or ALK rearranged cancers.
The authors conducted this study to evaluate the association of KRAS status with outcomes following ICI monotherapy versus chemoimmunotherapy in patients with PD-L1 of 50% or greater. The researchers used the Flatiron Health database, comprising 280 cancer clinics across the US and analyzed 1127 patients with advanced non-squamous NSCLC with PD-L1 expression of 50% or greater, known KRAS variant status, and no alteration in EGFR, ALK, or ROS1, who were treated with first line ICI monotherapy or chemoimmunotherapy between January 2016 and May 2020. Of the patients analyzed, 50.8% had KRAS variant status and 49.2% had KRAS wild type status. Patients with KRAS variant status were more likely to be female (58.7% versus 47.1%; P =0.002) and had smoking history (96.4% versus 87.7%; P < .001). Other patient demographics and patient characteristics, including age, race, ethnicity, Performance Status, and stage at diagnosis, were well balanced among the groups analyzed. Patient groups were stratified by treatment type and KRAS status (variant or wild type), and Overall Survival (OS) was compared between the treatment groups. Adjusted Hazard ratios for death associated with KRAS status and treatment regimen was estimated, using Cox proportional hazards models.
It was noted that among patients treated with ICI monotherapy, KRAS variant status was associated with superior median survival compared with KRAS wild type (21.1 months versus 13.6 months; HR=0.77; P=0.03), and this was statistically significant. However, among patients treated with chemoimmunotherapy, there was no significant median survival difference between patients with KRAS variant and KRAS wild type status (20.0 months versus 19.3 months; HR=0.99; P=0.93).
Among patients with KRAS variant status, the median OS did not differ between those treated with ICI monotherapy and chemoimmunotherapy (21.1 months versus 20.0 months; P =0.78), whereas among patients with KRAS wild type status, those treated with ICI monotherapy had numerically worse median survival than those treated with chemoimmunotherapy, although this difference was not statistically significant (13.6 months versus 19.3 months; HR=1.19; P =0.06).
In conclusion, this data suggests that chemoimmunotherapy might be favored over ICI monotherapy for patients with KRAS wild type tumors associated with high PD-L1 expression. The authors caution that in this analysis KRAS variant subtype and co-mutation status including TP53 and STK11 was unknown, and further investigation is needed to selection appropriate therapies for patients with PD-L1 High NSCLC.
Association Between KRAS Variant Status and Outcomes With First-line Immune Checkpoint Inhibitor–Based Therapy in Patients With Advanced Non–Small-Cell Lung Cancer. Sun L, Hsu M, Cohen RB, et al. JAMA Oncol. 2021;7:937-939.
Stereotactic Ablative Radiotherapy Non-Inferior to Surgery in Operable Stage I Non Small Cell Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Approximately 15% of patients present with early stage (T1-2 N0) disease, and these numbers are likely to increase with the implementation of Lung Cancer screening programs. Patients with early stage disease unless medically unfit, undergo surgical resection with a curative intent. Those who are not surgical candidates are often treated with conventional Radiation Therapy, over a period of 4 to 6 weeks.
Dating back to the 1930’s, the only hope for curing lung cancer has been surgery. However, important advances in the field of medical physics, computer science, and engineering have enabled significant progress in the field of Radiation Oncology, by better targeting the tumor and escalating the daily treatment doses. Surgery for Stage I NSCLC is now being challenged by these new Radiation Therapy techniques.
Stereotactic Ablative Radiotherapy (SABR) is a non-surgical procedure that allows delivery of significantly higher doses of precisely focused radiation to the tumor, compared to conventional Radiation Therapy, with less collateral damage to the surrounding normal tissue. The technologies used for SABR include GAMMA KNIFE® which uses highly focused gamma rays, Proton Beam therapy which uses ionized Hydrogen or Protons, Linear Accelerator (LINAC) and CYBER KNIFE® which use Photons, to target the tumor tissue. Because SABR is fractionated and delivered over 1-5 days, the short-and long-term side effects of radiation therapy are decreased and may allow higher total dosage to be given.
In a previously published pooled analysis of two independent, randomized, Phase III trials of SABR in patients with operable, clinical T1–2a (<4 cm), N0M0, Stage I NSCLC (STARS and ROSEL), Overall Survival (OS) was higher after Stereotactic Ablative Radiotherapy (SABR) than with surgery. This analysis had notable limitations and was closed early due to slow accrual. In the present study, the SABR group in the STARS trial was re-accrued with a larger sample size and the authors reported long-term results of the revised STARS trial, along with a protocol-specified propensity-matched comparison with a prospectively registered, contemporary institutional cohort of patients, who underwent Video-Assisted Thoracoscopic Surgical Lobectomy with Mediastinal Lymph Node Dissection (VATS L-MLND).
This single-arm prospective trial done at the University of Texas MD Anderson Cancer Center did not include patients from the previous pooled analysis and enrolled 80 patients (N=80) with newly diagnosed and histologically confirmed NSCLC with N0M0 disease (squamous cell, adenocarcinoma, large cell, or NSCLC not otherwise specified), and a tumor diameter of 3 cm or less. SABR dosing for peripheral lesions was 54 Gy in three fractions and 50 Gy in four fractions for central tumors, with simultaneous integrated boost to gross tumor totaling 60 Gy.
For the propensity-matching analysis, the researchers used a surgical cohort from the MD Anderson Department of Thoracic and Cardiovascular Surgery's prospectively registered, institutional review board-approved database of all patients with clinical Stage I NSCLC who underwent VATS L-MLND during the period of enrolment in this trial. Propensity matching consisted of determining a propensity score using a several covariates such as age, tumor size, histology, Performance Status, and the interaction of age and sex. The Primary endpoint was the 3-year Overall Survival. Non-inferiority could be claimed if the 3-year Overall Survival rate after SABR was lower than that after VATS L-MLND by 12% or less and the upper bound of the 95% CI of the Hazard Ratio (HR) was less than 1.965.
At a median follow-up time was 5.1 years, the OS with SABR was 91% at 3 years and 87% at 5 years. The OS in the propensity-matched VATS L-MLND cohort was 91% at 3 years and 84% at 5 years. Non-inferiority was claimed since the 3-year OS after SABR was not lower than that observed in the VATS L-MLND group. There was no significant difference in OS between the two patient cohorts from a multivariable analysis (HR=0.86; P=0•65). SABR was well tolerated with no Grade 4-5 toxicities.
It was concluded from this study that long term survival after SABR is non-inferior to VATS L-MLND for operable Stage IA Non Small Cell Lung Cancer. SABR remains promising for this patient group and the authors strongly recommend a multidisciplinary management approach .
Stereotactic ablative radiotherapy for operable stage I non-small-cell lung cancer (revised STARS): long-term results of a single-arm, prospective trial with prespecified comparison to surgery. Chang JY, Mehran RJ, Feng L, et al. Lancet Oncol. 2021;22:1448-1457.
Sintilimab for Patients with Pretreated EGFR-Mutated Non Small Cell Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR mutations and 90% of these mutations are either exon 19 deletions or L858R substitution mutation in exon 21. Patients with advanced EGFR-mutated NSCLC, following initial clinical response to first, second and third generation EGFR-TKIs therapies, will inevitably advance to a progressive disease course. These patients often receive platinum-based chemotherapy, with limited clinical benefit. Immune checkpoint inhibitors given alone have low efficacy in the treatment of patients with metastatic NSCLC with oncogenic-driven tumors. There is a highly unmet medical need for these patients with resistant disease.
Sintilimab is an immunoglobulin G4, anti-PD-1 monoclonal antibody, which binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. By doing so, it unleashes the tumor-specific effector T cells, and is thereby able to undo PD-1 pathway-mediated inhibition of the immune response.
ORIENT-31 is a prospective, randomized, double-blind, multi-center Phase III study, which evaluated Sintilimab, with or without a Bevacizumab biosimilar injection (IBI305), in combination with chemotherapy (Pemetrexed and Cisplatin), in patients with EGFR-mutated locally advanced or metastatic non-squamous NSCLC, who have progressed following EGFR TKI treatment. Patients were randomized in a 1:1:1 ratio to receive Sintilimab 200 mg IV plus Bevacizumab biosimilar 15 mg/kg IV combined with Pemetrexed 500 mg/m2 IV and Cisplatin 75 mg/m2 IV (Arm A), Sintilimab combined with Pemetrexed and Cisplatin (Arm B), or chemotherapy alone with Pemetrexed and Cisplatin (Arm C), all agents administered every 3 weeks for 4 cycles followed by maintenance treatment with Sintilimab plus Bevacizumab and Pemetrexed in Arm A, Sintilimab and Pemetrexed in Arm B, and Pemetrexed alone in Arm C. Treatment was continued until radiographic disease progression or unacceptable toxicity. Eligible patients included patients with disease progression following first or second generation EGFR TKI and confirmed as T790M negative, or T790M positive but further progressed on third generation EGFR TKI, or patients with disease progression following third generation EGFR TKI as first line treatment. The median age was 57 years, 36% of patients had brain metastasis, 64% of patients had received First or Second generation TKIs without T790M mutation, 28% had received First or Second generation TKIs and then a Third generation TKI for T790M mutation, and 8% patients received first line Third generation TKI. The target accrual was 480 patients and by the data cutoff date of the first interim analysis, 444 patients were enrolled. The Primary endpoint was Progression Free Survival (PFS) as assessed by an Independent Radiographic Review Committee (IRRC). Secondary endpoints included Overall Survival (OS), PFS as assessed by investigators, Objective Response Rate (ORR) and Safety. The median follow up at first interim analysis was 9.8 months.
Sintilimab plus Bevacizumab biosimilar in combination with chemotherapy (Arm A), demonstrated a statistically significant and clinically meaningful improvement in PFS, compared with Arm C (chemotherapy alone group). The median PFS was 6.9 months in Arm A, and 4.3 months in Arm C (HR=0.46; P<0.0001). Additionally, the key Secondary endpoints of ORR and Duration of Response (DOR) were improved in Arm A compared with Arm C, and the results of PFS, ORR and DOR assessed by the investigator were consistent with the results assessed by IRRC. The prespecified PFS futility analysis that compares Arm A to Arm B (Sintilimab and chemotherapy group) did not cross futility stopping boundary. The PFS data of Arm B versus Arm C were immature.
The authors concluded that in this first prospective, double-blind, Phase III study among patients with EGFR mutated NSCLC who had progressed after EGFR TKIs, this quadruple regimen of Sintilimab plus Bevacizumab biosimilar in combination with chemotherapy, significantly improved Progression Free Survival, compared with chemotherapy alone.
VP9-2021: ORIENT-31: Phase III study of sintilimab with or without IBI305 plus chemotherapy in patients with EGFR mutated nonsquamous NSCLC who progressed after EGFR-TKI therapy. Lu S, Wu L, Jian H, et al. Published:November 19, 2021DOI:https://doi.org/10.1016/j.annonc.2021.10.007.
Adherence to Guideline-Recommended Biomarker Testing as an Integral Component of NSCLC Care
Written by: Dr. David M. Waterhouse, M.D., MPH
Content Sponsored by: Amgen
The NSCLC Landscape Has Evolved Significantly Due Largely to the Growing Number of Actionable Mutations1
Despite innovations in standard-of-care, advanced non-small cell lung cancer (NSCLC) continues to burden patients, with poor survival outcomes.2,3 NSCLC has been identified as the leading cause of cancer death worldwide with an estimated 1.8 million deaths in 2020.2 As the number of targeted therapies and approval companion diagnostics continues to grow, mortality and survival rates have begun to improve.3 With the addition of KRAS G12C, there are 9 actionable molecular biomarkers (as of October 2021) and more than 20 targeted therapies approved for use in advanced NSCLC.1,4 Guidelines recommend biomarker testing for all eligible patients at diagnosis of advanced NSCLC regardless of characteristics such as smoking history, race, or sex.5,6 Unfortunately, real-world evidence shows that far too many patients fail to receive comprehensive biomarker testing, despite their eligibility.7,8
Adherence to Guidelines Can Improve Biomarker Testing9
As targeted therapies are approved, guidelines continue to update their recommendations on biomarker testing.5 As of September 2021, NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC recommend broad molecular testing of actionable and emerging biomarkers for patients with advanced or metastatic NSCLC (Figure 1).5 Similarly, the American Society of Clinical Oncology (ASCO) endorsed the 2018 College of American Pathologists (CAP)/International Association for the Study of Lung Cancer (IASLC)/Association for Molecular Pathology (AMP) guidelines, recommending comprehensive cancer panel testing for genetic biomarkers.10,11
Figure 1: NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC5,*,†
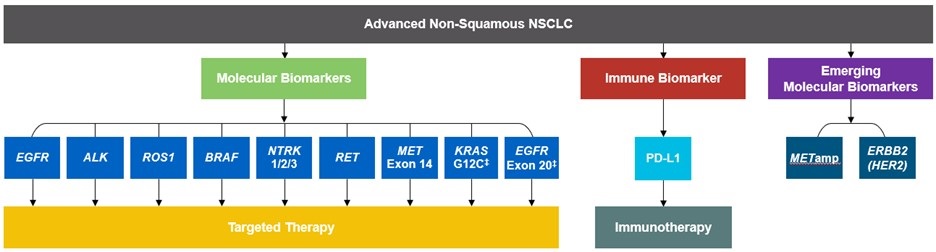
*The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC provide recommendations for certain individual biomarkers that should be tested and recommend testing techniques but do not endorse any specific commercially available biomarker assays or commercial laboratories.5 †)The NCCN Guidelines® for NSCLC recommend broad molecular testing to identify rare driver variants for which targeted therapies may be available to ensure patients receive the most appropriate treatment.5‡KRAS G12C and EGFR exon 20 mutations are used to determine subsequent (ie, second-line and beyond) therapy using targeted agents or other novel agents.5
Although adherence to guideline-recommended biomarker testing is associated with improved patient outcomes, real-world EMR data reveals suboptimal biomarker testing rates.7,9 In a retrospective study, 81% of patients with metastatic NSCLC did not receive testing for ALK, EGFR, ROS1, and BRAF before initiation of first-line treatment, despite the availability of targeted therapies.7 Moreover, only 28% of patients received testing for all four genetic biomarkers and PD-L1 during the study period.7 In another retrospective study, less than 50% of patients with metastatic NSCLC received testing for all five biomarkers (EGFR, ALK, ROS1, BRAF, PD-L1) (Figure 2).8
Beyond the underutilization of biomarker testing, there remains an even greater need to increase broad molecular testing among racial and ethnic minority groups in the US.12,13 In one retrospective study, Black/African American patients with advanced NSCLC had significantly lower rates of testing with NGS assays (43.8%) compared with White patients (54.7%) (Figure 3).12
Figure 2: MYLUNG Consortium™ EMR Analysis of Patients With Metastatic NSCLC8,§
§A retrospective, observational study assessing real-world biomarker testing patterns in patients with metastatic NSCLC from community oncology practices within the US Oncology Network community practices between 2018 and 2020.8
Figure 3: EMR Analysis of Biomarker Testing in Patients With Advanced/Metastatic NSCLC12,**
**A retrospective cohort study of patients with advanced/metastatic non-squamous NSCLC (N=10,333) from ~ 800 sites of care in the US identified via the Flatiron Electronic Health Record Database between 2017 and 2020.12
Collectively, these findings highlight the disparity in proactive disease management across different patient populations.7,8,12
Considerations Across the Biomarker Testing Journey
There are several different methods in which eligible patients can be tested for actionable genetic alterations, each with unique considerations as indicated below (Figure 4).
Figure 4: Comparing Biomarker Testing Methods and Sample Types1,6,11,14,15
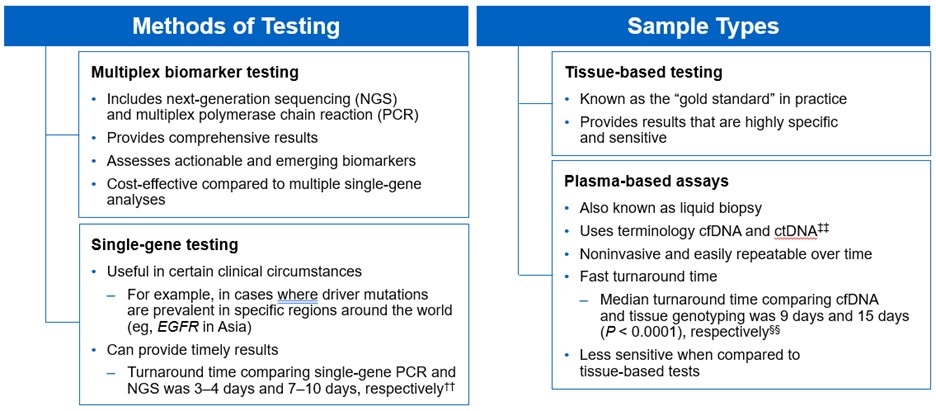
‡‡Data from a review of common molecular assays for biomarker testing that analyzed common detected variants, sensitivities, and turnaround time.6 ‡‡cfDNA refers to all circulating DNA (largely non-malignant), while ctDNA refers to the tumor-related component of cfDNA.14 §§Data from a prospective study that enrolled patients with previously untreated metastatic NSCLC undergoing SOC tissue genotyping and comprehensive cfDNA analysis, with turnaround time defined as the number of days between the test order date and the retrieval of test results.15
While tissue biopsy remains the “gold standard” in NSCLC, it may not be feasible (insufficient tissue) or pragmatic (urgent need to begin treatment) in all patients.6,14,16,17 Plasma ctDNA demonstrates complementary results to tissue-based assays and can be considered a valid tool for genotyping of newly diagnosed patients with advanced NSCLC.14 In a prospective study from 2016 to 2018, guideline-recommended biomarkers with FDA-approved therapies (EGFR Exon 19 deletion and L858R, ALK fusion, ROS1 fusion, BRAF V600E) showed ≥ 98.2% concordance between tissue and liquid-based testing.15 While concordance is high for any single test, high concordance for full panels will be required for liquid biopsies to become standard. Additionally, negative results on liquid biopsy still require validation with tissue testing.14,15,17
Liquid biopsy offers improvements in sample acquisition and small tissue samples and provides less invasive procedures and shortened turnaround times.14,15,18 Other considerations for maximizing the tissue journey include the use of comprehensive testing, rapid on-site evaluation (ROSE), and implementing reflex testing protocols with the help of a multidisciplinary team (MDT).6,17,18
Delays in Biomarker Testing Results May Impact Treatment Decisions16
Longer turnaround times for molecular testing compared with turnaround times for PD-L1 testing by IHC may result in the initiation of immunotherapy before molecular testing results are received.16 Waiting for complete biomarker test results prior to initiating therapy can allow doctors to make the most informed decisions surrounding a patient’s treatment journey.16 The benefits of delaying therapy may outweigh the risks associated with initiating immunotherapy treatment in patients with actionable genetic alterations who are eligible for targeted treatments, such as immunotherapy treatments, which are associated with a delay in benefit when compared to targeted therapy.16
Ensuring Best Practices in Biomarker Testing Can Lead to Better Patient Outcomes9
As the NSCLC landscape continues to progress with the increasing number of actionable biomarkers, there is a growing need for proactive and comprehensive molecular testing.1,7,8,12,17 Although real-world data has shown significant underuse of biomarker testing, rates can be improved with a diligent observation of expanding guidelines and recommendations by expert panels and associations.7-9,12 In the coming years, it will be increasingly urgent for clinicians to evolve institutional protocols, including enabling reflex testing, and work as an MDT to ensure biomarker testing is performed on all eligible patients with advanced NSCLC.6,17,18
[Abbreviations]
ALK, anaplastic lymphoma kinase; BRAF, proto-oncogene B-Raf; cfDNA, cell-free DNA;
ctDNA, circulating tumor DNA; EGFR, epidermal growth factor receptor; EMR, electronic medical record; ERBB2, erb-b2 receptor tyrosine kinase 2; HER2, human epidermal growth factor receptor 2;
IHC, immunohistochemistry; KRAS, Kirsten rat sarcoma viral oncogene homolog; MET, mesenchymal-to-epithelial transition; mNSCLC, metastatic non-small cell lung cancer; NCCN, National Comprehensive Cancer Network; NTRK, neurotrophic tyrosine receptor kinase; PD-L1, programmed cell death ligand 1; RET, rearranged during transfection; ROS1, c-ros oncogene 1; SOC, standard-of-care.
[References]
1. Majeed U, et al. J Hematol Oncol. 2021;14:108.
2. Sung H, et al. CA Cancer J Clin. 2021;71:209-249.
3. Siegel RL, et al. CA Cancer J Clin. 2021;71:7-33.
4. Food and Drug Administration. www.fda.gov. Accessed October 6, 2021.
5. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer. v.6.2021. ©National Comprehensive Cancer Network, Inc. 2021. All rights reserved. Accessed October 8, 2021. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
6. Pennell NA, et al. Am Soc Clin Oncol Educ Book. 2019;39:531-542.
7. Nadler ES, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 9079.
8. Robert NJ, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 102.
9. John A, et al. Adv Ther. 2021;38:1552-1566.
10. Hanna N, et al. J Clin Oncol. 2017;35:3484-3515.
11. Lindeman NI, et al. Arch Pathol Lab Med. 2018;142:321-346.
12. Bruno DS, et al. Presented at: The American Society of Clinical Oncology Annual Meeting; June 4–8, 2021; Virtual Meeting. Abstract 9005.
13. Hann KEJ, et al. BMC Public Health. 2017;17:503.
14. Rolfo C, et al. J Thorac Oncol. 2021;16:1647-1662.
15. Leighl NB, et al. Clin Cancer Res. 2019;25:4691-4700.
16. Smeltzer MP, et al. J Thorac Oncol. 2020;15:1434-1448.
17. Gregg JP, et al. Transl Lung Cancer Res. 2019;8:286-301.
18. VanderLaan PA, et al. Cancer Cytopathol. 2020;128:629-636.
USA-510-80864 10/21
FDA Approves TECENTRIQ® as Adjuvant Treatment for Non Small Cell Lung Cancer
SUMMARY: The FDA on October 15, 2021, approved TECENTRIQ® (Atezolizumab) for adjuvant treatment, following resection and Platinum-based chemotherapy, in patients with Stage II to IIIA Non-Small Cell Lung Cancer (NSCLC) whose tumors have PD-L1 expression on 1% or more of tumor cells, as determined by an FDA-approved test. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Surgical resection is the primary treatment for approximately 30% of patients with NSCLC who present with early Stage (I–IIIA) disease. These patients are often treated with platinum-based adjuvant chemotherapy to decrease the risk of recurrence. Nonetheless, 45-75% of these patients develop recurrent disease. There is therefore an unmet need for this patient population.
TECENTRIQ® is an anti PD-L1 monoclonal antibody, designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors expressed on activated T cells. PD-L1 inhibition may prevent T-cell deactivation and further enable the activation of T cells.
IMpower 010 is a global, multicentre, open-label, randomized Phase III study evaluating the efficacy and safety of TECENTRIQ® compared with Best Supportive Care (BSC), in patients with Stage IB-IIIA NSCLC, following surgical resection and up to 4 cycles of adjuvant Cisplatin-based chemotherapy. In this study, 1005 patients were randomized 1:1 to receive TECENTRIQ® 1200 mg IV every 3 weeks for 16 cycles, or BSC. Both study groups were well balanced and eligible patients had an ECOG PS of 0-1. The Primary endpoint was Disease Free Survival (DFS) in the PD-L1-positive Stage II-IIIA patients, all randomized Stage II-IIIA patients and Intent to Treat (ITT) Stage IB-IIIA populations. Key Secondary endpoints included Overall Survival (OS) in the overall study population and ITT Stage IB-IIIA NSCLC patients. At data cutoff on January 21, 2021, median follow up was 32.2 months in the ITT population.
Treatment with TECENTRIQ® following surgery and chemotherapy reduced the risk of disease recurrence or death (DFS-Disease Free Survival) by 34% (HR=0.66; P=0.0039), in patients with Stage II-IIIA NSCLC, whose tumor PD-L1 expression was 1% or more, compared with BSC. In this patient population, median DFS was Not Reached for TECENTRIQ®, compared with 35.3 months for BSC. This benefit was even more so among Stage II-IIIA NSCLC patients with PD-L1 expression 50% or more. Adjuvant TECENTRIQ® following surgery and chemotherapy in this patient group reduced the risk of disease recurrence or death (DFS) by 57% (HR=0.43). In the larger population of all randomized Stage II-IIIA study patients, TECENTRIQ® reduced the risk of disease recurrence or death by 21% (HR=0.79, P=0.02). In this patient population, TECENTRIQ® increased DFS by a median of seven months, compared with BSC (42.3 months versus 35.3 months). The significance boundary was not crossed for DFS in the ITT patient population. Overall Survival data were immature and not formally tested. Safety data for TECENTRIQ® were consistent with its known safety profile and no new safety signals were identified.
It was concluded that this study met its Primary endpoint, and is the first Phase III study to demonstrate that treatment with TECENTRIQ® following surgery and chemotherapy can significantly delay disease recurrence in patients with early stage lung cancer, with a more pronounced benefit noted, in patients with tumor PD-LI expression of 1% or more.
IMpower010: Primary results of a phase III global study of atezolizumab versus best supportive care after adjuvant chemotherapy in resected stage IB-IIIA non-small cell lung cancer (NSCLC). Wakelee HA, Altorki NK, Zhou C, et al. J Clin Oncol. 2021;39:(suppl 15; abstr 8500). doi:10.1200/JCO.2021.39.15_suppl.8500
Durable Survival Benefit with First Line OPDIVO® plus YERVOY® and a Limited Course of Chemotherapy
SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the immune system T cells. Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152) and PD-1(Programmed cell Death 1). Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response. OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4. In the CheckMate-227, Part 1, Phase III trial, a combination of OPDIVO® plus YERVOY®, significantly improved Overall Survival (OS), Progression Free Survival (PFS), Objective Response Rates (ORR) and Duration of Response, compared to chemotherapy, independent of PD-L1 expression level. The authors in this study hypothesized that a limited course of chemotherapy combined with OPDIVO® plus YERVOY® could provide rapid disease control, while building on the durable Overall Survival benefit seen with dual PD-1 and CTLA-4 inhibition, as well as minimizing the toxicities associated with a full course of chemotherapy.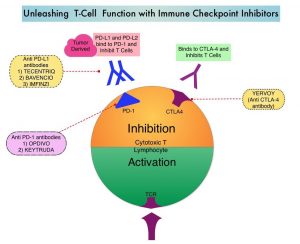
CheckMate-9LA is a randomized, open-label, multi-center, Phase III trial which evaluated the benefit of a combination of OPDIVO® plus YERVOY®, and 2 cycles of Platinum-doublet chemotherapy (experimental arm) versus Platinum-doublet chemotherapy (control arm) for 4 cycles, followed by optional Pemetrexed maintenance therapy, as a first-line treatment in patients with metastatic or recurrent NSCLC, regardless of PD-L1 status and histology. In this study, 719 adults treatment naïve patients with histologically confirmed Stage IV/recurrent NSCLC, with ECOG Performance Status 0-1, and no known sensitizing EGFR/ALK alterations, were randomly assigned 1:1 to receive OPDIVO® 360 mg every 3 weeks plus YERVOY® 1 mg/kg every 6 weeks and 2 cycles of platinum-doublet chemotherapy (N=361), or 4 cycles of platinum-doublet chemotherapy alone (N=358). Chemotherapy was based on histology. Patients with non-squamous NSCLC in the chemo-only randomized group could receive optional Pemetrexed maintenance treatment. Patients were treated with immunotherapy until disease progression, unacceptable toxicity, or for 2 years. Patients were stratified by PD-L1 status (less than 1% versus 1% or more), sex, and histology (squamous versus non-squamous). Demographics in treatment groups were well balanced. Crossover between treatment groups was not permitted. However, at physician discretion, patients could receive subsequent immunotherapy upon discontinuation of study treatment in either group.
The Primary end point was Overall Survival (OS). Secondary endpoints included Progression Free Survival (PFS), Objective Response Rate (ORR) and efficacy by PD-L1 subgroups. PFS2 was a pre-specified exploratory endpoint and was defined as time from randomization, to objectively documented progression after the next line of therapy, or to death from any cause, whichever occurred first. At a preplanned interim analysis after a minimum follow up 8.1 months, this trial met its primary and secondary endpoints, showing statistically significant improvements in OS, PFS, and Objective Response Rate (ORR), when compared to chemotherapy alone. This clinical benefit was noted across tumor PD-L1 expression levels and histologies.
The authors in this publication reported updated efficacy and safety outcomes, along with Progression-Free Survival (PFS) after next line of treatment (PFS2), Treatment-Related Adverse Events (TRAEs) by treatment cycle, and efficacy outcomes in patients who discontinued all treatment components in the experimental treatment group due to TRAEs, from the CheckMate 9LA Phase III trial. The minimum follow up for OS was 24.4 months. The majority of patients (93%) received two cycles of chemotherapy and 13% completed the maximum 2 years of immunotherapy treatment. The median number of doses was 9.0 for OPDIVO® and 4.0 for YERVOY®. In the control arm, 75% of patients received four cycles of chemotherapy and 67% patients who had non-squamous tumor histology receiving Pemetrexed maintenance. About 29% patients in the control arm had completed the full four cycles of chemotherapy without optional Pemetrexed maintenance therapy. The median duration of therapy was 6.1 months in the experimental arm and 2.5 months in the control arm.
With a median follow up of 30.7 months, OPDIVO® plus YERVOY® with a limited course of chemotherapy continued to prolong Overall Survival (OS), when compared to chemotherapy (Median OS 15.8 versus 11.0 months; HR=0.72). The 2-year OS rate was 38% versus 26%. This OS benefit was observed across most key subgroups including those with PD-L1 expression of less than 1%, more than 1%, as well as by histology. More importantly, patients with pretreated CNS metastases at baseline had a median OS of 19.9 months in the experimental group versus 7.9 months in the control group, respectively (HR=0.47).
PFS continued to be prolonged in the experimental group compared to the control group, with an Hazard Ratio of 0.67 and 2-year PFS rates of 20% versus 8%, respectively. The ORR was 38% in the experimental group and 25% in the control group (P=0.0003). 34% versus 12% of all responses respectively, were ongoing at 2 years. The median PFS2 in all randomized patients was 13.9 months in the experimental group and 8.7 months in the control group (HR=0.66). Again, PFS2 also favored the experimental arm over the control arm in subgroups by PD-L1 expression, and by histology.
No new safety signals were observed and majority of Grade 3/4 toxicities were mostly observed during the first two treatment cycles in the experimental group. In patients who discontinued all components of the experimental treatment (OPDIVO® plus YERVOY® with chemotherapy) due to toxicities (N=61), the median OS was 27.5 months and 56% of responders had an ongoing response, more than 1 year after discontinuation of therapy. After discontinuing the experimental regimen, patients remained treatment-free for a median of 11.9 months and had a 48% chance of being treatment-free at 1 year.
The researchers concluded that with a 2-year minimum follow-up, OPDIVO® plus YERVOY® with two cycles of chemotherapy provided durable efficacy benefits over conventional chemotherapy, with a manageable safety profile. They added that this treatment regimen remains an efficacious first line treatment of advanced Non Small Cell Lung Cancer.
First-line nivolumab plus ipilimumab with two cycles of chemotherapy versus chemotherapy alone (four cycles) in advanced non-small-cell lung cancer: CheckMate 9LA 2-year update. Reck M, Ciuleanu T-E, Cobo M, et al. https://doi.org/10.1016/j.esmoop.2021.100273
FDA Approves EXKIVITY® for Metastatic Non Small Cell Lung Cancer with EGFR exon 20 Insertion Mutations
SUMMARY: The FDA on September 15, 2021, granted accelerated approval to EXKIVITY® (Mobocertinib), for adult patients with locally advanced or metastatic Non Small Cell Lung Cancer (NSCLC) with Epidermal Growth Factor Receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after Platinum-based chemotherapy. The FDA also approved the Oncomine Dx Target Test as a companion diagnostic device to select patients with the above mutations for EXKIVITY® treatment.
The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR mutations and 90% of these mutations are either exon 19 deletions or L858R substitution mutation in exon 21. EGFR exon 20 insertion mutations are the third most common after L858R and exon 19 deletions, and occur in about 2-3% patients with NSCLC, and are insensitive to EGFR Tyrosine Kinase Inhibitors (TKIs) due to an altered conformation of the kinase active site. Next-Generation sequencing provides an alternative to Polymerase Chain Reaction (PCR)-based tests, which fail to identify 50% or more of exon 20 insertion mutations. Patients with EGFR exon 20 insertion mutations have a 5 year Overall Survival (OS) of 8% in the frontline setting, compared to an OS of 19% for patients with EGFR exon 19 deletions or L858R mutations. There is therefore a clinically unmet need for this patient group, as there are no approved targeted therapies available and platinum-doublet chemotherapy remains the standard of care for these patients.
EXKIVITY® is a novel oral EGFR TKI, that was designed to address the unmet need in patients with EGFR exon 20 insertion mutant positive NSCLC. EXKIVITY® was designed to potently inhibit oncogenic variants containing activating mutations in exon 20, with selectivity over Wild Type EGFR.
The present FDA approval of EXKIVITY® was based on an international, non-randomized, open-label, multicohort clinical trial (NCT02716116) which included patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. This trial was conducted in three parts- Dose escalation or Part 1, to determine the safety profile of EXKIVITY®, Expansion phase or Part 2, to evaluate the antitumor activity of EXKIVITY® in seven histologically and molecularly defined cohorts, and Extension phase or Part 3, to evaluate efficacy of EXKIVITY® in patients with locally advanced or metastatic NSCLC whose tumors harbor EGFR exon 20 insertion mutations, and who have been previously treated.
So, this 3-part, multicenter study has dose-escalation/expansion and extension (EXCLAIM) cohorts. Patients with EGFR exon 20 insertion positive metastatic NSCLC, with ECOG status 0-1, and one or more prior lines of therapy for locally advanced/metastatic disease, received EXKIVITY® 160 mg orally daily, until disease progression or intolerable toxicity. Efficacy was evaluated in 114 patients whose disease had progressed on or after Platinum-based chemotherapy. Among these platinum pretreated patients, the median age was 60 years, 66% were female, 60% were Asian, and 59% had 2 or more prior systemic therapies. The main efficacy outcome measures were confirmed Objective Response Rate (ORR) assessed by blinded Independent Review Committee (IRC) and Duration of Response.
At a median follow up of 14.2 months, the ORR was 28%, with a median Duration of Response of 17.5 months. The median Progression Free Survival was 7.3 months and the median Overall Survival was 24 months. Clinically meaningful improvements were observed for dyspnea, coughing, chest pain, and these benefits were evident by cycle 2, and maintained throughout treatment. The most common adverse reactions were rash, nausea, stomatitis, vomiting, diarrhea, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain.
It was concluded that EXKIVITY® is the first and only oral therapy specifically designed to target EGFR exon 20 insertions, and the present FDA approval is an important addition, designed for this patient population.
Mobocertinib (TAK-788) in EGFR exon 20 insertion (ex20ins)+ metastatic NSCLC (mNSCLC): additional results from platinum-pretreated patients (pts) and EXCLAIM cohort of phase 1/2 study. Ramalingam SS, Zhou C, Kim TM, et al. J Clin Oncol. 2021;39(suppl 15):9014. DOI:10.1200/JCO.2021.39.15_suppl.9014.
Real-world evidence: What can it inform us about the second-line treatment of metastatic squamous NSCLC?
Written by Dr. Solly S. Chedid
Sponsored and developed by Boehringer Ingelheim Pharmaceuticals.
Immunotherapies have changed the way we initiate treatment for many patients with advanced squamous non-small cell lung cancer (NSCLC).1 As immunotherapy has become a standard first-line treatment, non-immunotherapy options are important to consider for second-line treatment. Currently, there is no clear standard of care for second-line therapy in patients with advanced squamous NSCLC who progress after immuno-chemotherapy. Therefore, an unmet need remains for studies designed to understand the effectiveness and safety of second-line treatments in these patients. Here we will review newly published real-world evidence on second-line treatments of patients with squamous NSCLC with afatinib (GILOTRIF®) following immuno-chemotherapy.
GILOTRIF is the only oral, chemotherapy-free option for treating patients with squamous NSCLC that has progressed after platinum-based chemotherapy.2 The efficacy and safety of GILOTRIF were demonstrated in the pivotal LUX-Lung 8 trial. In LUX-Lung 8, treatment with GILOTRIF led to statistically significant improvements in progression-free survival (median 2.4 vs 1.9 months) and overall survival (median 7.9 vs 6.8 months) compared with erlotinib. In LUX-Lung 8, the most common adverse reactions reported in the GILOTRIF treated patients (≥20% all grades) were diarrhea (75%), rash/acneiform dermatitis (70%), stomatitis (30%), decreased appetite (25%), and nausea (21%).
The Real-world Effectiveness of 2L Treatment of Squamous mNSCLC Study is the first to evaluate the real-world use of GILOTRIF following first-line immuno-chemotherapy in patients with squamous NSCLC.1 It is a retrospective, non-interventional, multisite cohort study using electronic medical records of patients with advanced or metastatic squamous NSCLC treated with pembrolizumab and platinum-doublet chemotherapy in the first line. Patients were treated with either GILOTRIF or physician’s choice chemotherapy in the second line. Study endpoints included patient demographics and clinical characteristics, time on second-line treatment, and incidence of severe (Grade ≥3) immune-related adverse events (irAEs). This study analysis was not powered to compare characteristics or outcomes between the cohorts. In addition, the results of this study are not intended for direct comparison with clinical trials. The main limitations of this study are its retrospective nature, potential for selection bias, and lack of a comparator arm.
A total of 200 patients were included in this study; 99 received GILOTRIF, and 101 received chemotherapy in the second line.1 More patients in the GILOTRIF cohort had mixed histology, were epidermal growth factor receptor (EGFR) mutation−positive, and were never smokers than those in the chemotherapy cohort. There were geographic differences between the cohorts; more patients from the Northeast received GILOTRIF, and more patients from the South received chemotherapy. In the GILOTRIF cohort, 45% of patients had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) ranging from 0 to 1, while 55% had an ECOG PS of 2 or higher. In the chemotherapy arm, 50% of patients had ECOG PS 0 to 1, and 50% had ECOG PS of 2 or higher. Other characteristics, such as median age and stage at diagnosis, were similar in both cohorts.
The median time on treatment for the GILOTRIF cohort was 7.3 months. In patients with mixed histology, the median time on treatment was 8.1 months, and for patients with squamous histology it was 5.8 months.1 EGFR mutation−positive and EGFR mutation−negative patients remained on GILOTRIF for a median of 7.4 and 5.9 months, respectively. The median time on treatment from initiation of second-line chemotherapy was 4.2 months.
Time on Treatment in the Real-World Effectiveness Study1

The most common adverse drug reactions with GILOTRIF were diarrhea (26%), rash (6%), stomatitis, fatigue, and nausea (5% each).1 Six out of 99 patients experienced a Grade 3/4 irAE during second-line GILOTRIF therapy; each of these patients also experienced a Grade 3 irAE during first-line treatment. The 6 patients in the GILOTRIF cohort who experienced Grade 3/4 irAEs were treated with steroids, and none were hospitalized. Given the real-world nature of the study, adverse event data may be underreported or underdocumented; in addition, censoring may also bias results.
Such real-world evidence (RWE) studies have limitations, including their retrospective nature and potential for selection bias.1 However, in addition to clinical data collected in registrational clinical trials, data from RWE studies such as this can add important information to help evaluate the clinical utility of a drug in the real-world setting.3 RWE studies can be derived from rich data sources, such as electronic health records, registries, and claims databases, which reflect real-world use, outcomes, and the patient diversity seen in clinical practice.
Despite several limitations highlighted in this paper, the study adds to the body of evidence supporting the effectiveness and safety of GILOTRIF when given as a second-line treatment following immuno-chemotherapy in routine clinical practice.1
INDICATION AND USAGE
GILOTRIF is indicated for the treatment of patients with metastatic squamous NSCLC progressing after platinum-based chemotherapy.
IMPORTANT SAFETY INFORMATION FOR GILOTRIF® (AFATINIB) TABLETS
WARNINGS AND PRECAUTIONS
Diarrhea
• GILOTRIF can cause diarrhea which may be severe and can result in dehydration with or without renal impairment. In clinical studies, some of these cases were fatal.
• For patients who develop Grade 2 diarrhea lasting more than 48 hours or Grade 3 or greater diarrhea, withhold GILOTRIF until diarrhea resolves to Grade 1 or less, and then resume at a reduced dose.
• Provide patients with an anti-diarrheal agent (e.g., loperamide) for self-administration at the onset of diarrhea and instruct patients to continue anti-diarrheal until loose stools cease for 12 hours.
Bullous and Exfoliative Skin Disorders
• GILOTRIF can result in cutaneous reactions consisting of rash, erythema, and acneiform rash. In addition, palmar-plantar erythrodysesthesia syndrome was observed in clinical trials in patients taking GILOTRIF.
• Discontinue GILOTRIF in patients who develop life-threatening bullous, blistering, or exfoliating skin lesions. For patients who develop Grade 2 cutaneous adverse reactions lasting more than 7 days, intolerable Grade 2, or Grade 3 cutaneous reactions, withhold GILOTRIF. When the adverse reaction resolves to Grade 1 or less, resume GILOTRIF with appropriate dose reduction.
• Postmarketing cases of toxic epidermal necrolysis (TEN) and Stevens Johnson syndrome (SJS) have been reported in patients receiving GILOTRIF. Discontinue GILOTRIF if TEN or SJS is suspected.
Interstitial Lung Disease
• Interstitial Lung Disease (ILD) or ILD-like adverse reactions (e.g., lung infiltration, pneumonitis, acute respiratory distress syndrome, or alveolitis allergic) occurred in patients receiving GILOTRIF in clinical trials. In some cases, ILD was fatal. The incidence of ILD appeared to be higher in Asian patients as compared to white patients.
• Withhold GILOTRIF during evaluation of patients with suspected ILD, and discontinue GILOTRIF in patients with confirmed ILD.
Hepatic Toxicity
• Hepatic toxicity as evidenced by liver function tests abnormalities has been observed in patients taking GILOTRIF. In 4257 patients who received GILOTRIF across clinical trials, 9.7% had liver test abnormalities, of which 0.2% were fatal.
• Obtain periodic liver testing in patients during treatment with GILOTRIF. Withhold GILOTRIF in patients who develop worsening of liver function. Discontinue treatment in patients who develop severe hepatic impairment while taking GILOTRIF.
Gastrointestinal Perforation
• Gastrointestinal (GI) perforation, including fatal cases, has occurred with GILOTRIF. GI perforation has been reported in 0.2% of patients treated with GILOTRIF among 3213 patients across 17 randomized controlled clinical trials.
• Patients receiving concomitant corticosteroids, nonsteroidal anti-inflammatory drugs (NSAIDs), or anti-angiogenic agents, or patients with increasing age or who have an underlying history of GI ulceration, underlying diverticular disease, or bowel metastases may be at an increased risk of perforation.
• Permanently discontinue GILOTRIF in patients who develop GI perforation.
Keratitis
• Keratitis has been reported in patients taking GILOTRIF.
• Withhold GILOTRIF during evaluation of patients with suspected keratitis. If diagnosis of ulcerative keratitis is confirmed, interrupt or discontinue GILOTRIF. If keratitis is diagnosed, the benefits and risks of continuing treatment should be carefully considered. GILOTRIF should be used with caution in patients with a history of keratitis, ulcerative keratitis, or severe dry eye. Contact lens use is also a risk factor for keratitis and ulceration.
Embryo-Fetal Toxicity
• GILOTRIF can cause fetal harm when administered to a pregnant woman. Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
• Advise females of reproductive potential to use effective contraception during treatment, and for at least 2 weeks after the last dose of GILOTRIF. Advise female patients to contact their healthcare provider with a known or suspected pregnancy.
ADVERSE REACTIONS
Adverse Reactions observed in clinical trials were as follows:
Previously Treated, Metastatic Squamous NSCLC
• In GILOTRIF-treated patients (n=392) the most common adverse reactions (≥20% all grades & vs erlotinib-treated patients (n=395)) were diarrhea (75% vs 41%), rash/acneiform dermatitis (70% vs 70%), stomatitis (30% vs 11%), decreased appetite (25% vs 26%), and nausea (21% vs 16%).
• Serious adverse reactions were reported in 44% of patients treated with GILOTRIF. The most frequent serious adverse reactions reported in patients treated with GILOTRIF were pneumonia (6.6%), diarrhea (4.6%), and dehydration and dyspnea (3.1% each). Fatal adverse reactions in GILOTRIF-treated patients included ILD (0.5%), pneumonia (0.3%), respiratory failure (0.3%), acute renal failure (0.3%), and general physical health deterioration (0.3%).
DRUG INTERACTIONS
Effect of P-glycoprotein (P-gp) Inhibitors and Inducers
• Concomitant use of P-gp inhibitors (including but not limited to ritonavir, cyclosporine A, ketoconazole, itraconazole, erythromycin, verapamil, quinidine, tacrolimus, nelfinavir, saquinavir, and amiodarone) with GILOTRIF can increase exposure to afatinib.
• Concomitant use of P-gp inducers (including but not limited to rifampicin, carbamazepine, phenytoin, phenobarbital, and St. John’s wort) with GILOTRIF can decrease exposure to afatinib.
USE IN SPECIFIC POPULATIONS
Lactation
• Because of the potential for serious adverse reactions in breastfed infants from GILOTRIF, advise women not to breastfeed during treatment with GILOTRIF and for 2 weeks after the final dose.
Females and Males of Reproductive Potential
• GILOTRIF may reduce fertility in females and males of reproductive potential. It is not known if the effects on fertility are reversible.
Renal Impairment
• Patients with severe renal impairment (estimated glomerular filtration rate [eGFR] 15 to 29 mL/min/1.73 m2) have a higher exposure to afatinib than patients with normal renal function. Administer GILOTRIF at a starting dose of 30 mg once daily in patients with severe renal impairment. GILOTRIF has not been studied in patients with eGFR <15 mL/min/1.73 m2 or who are on dialysis.
Hepatic Impairment
• GILOTRIF has not been studied in patients with severe (Child Pugh C) hepatic impairment. Closely monitor patients with severe hepatic impairment and adjust GILOTRIF dose if not tolerated.
GF PROF ISI 10.21.19
References
1. Kim ES, Kish JK, Cseh A, et al. Second-line afatinib or chemotherapy following immunochemotherapy for the treatment of metastatic, squamous cell carcinoma of the lung: real-world effectiveness and safety from a multisite retrospective chart review in the USA. Clin Lung Cancer. 2021;S1525-7304(21)00029-2.
2. GILOTRIF. Prescribing information. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc.; 2019.
3. Sherman RE, Anderson SA, Dal Pan GJ, et al. Real-world evidence — what is it and what can it tell us? N Engl J Med. 2016:8;375(23):2293-2297.
Please review the Full Prescribing Information at https://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Gilotrif/Gilotrif.pdf?DMW_FORMAT=pdf
Patient Information at https://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Gilotrif/Patient%20Info/gilotrif_patient%20information.pdf?DMW_FORMAT=pdf .
Adjuvant TECENTRIQ® Improves Disease Free Survival in Early Stage Non Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Surgical resection is the primary treatment for approximately 30% of patients with NSCLC who present with early Stage (I–IIIA) disease. These patients are often treated with platinum-based adjuvant chemotherapy to decrease the risk of recurrence. Nonetheless, 45-75% of these patients develop recurrent disease. There is therefore an unmet need for this patient population.
TECENTRIQ® (Atezolizumab) is an anti PD-L1 monoclonal antibody, designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors expressed on activated T cells. PD-L1 inhibition may prevent T-cell deactivation and further enable the activation of T cells.
IMpower 010 is a global, multicentre, open-label, randomized Phase III study evaluating the efficacy and safety of TECENTRIQ® compared with Best Supportive Care (BSC), in patients with Stage IB-IIIA NSCLC, following surgical resection and up to 4 cycles of adjuvant Cisplatin-based chemotherapy. In this study, 1005 patients were randomized 1:1 to receive TECENTRIQ® 1200 mg IV every 3 weeks for 16 weeks, or BSC. Both study groups were well balanced and eligible patients had an ECOG PS of 0-1. The Primary endpoint was Disease Free Survival (DFS) in the PD-L1-positive Stage II-IIIA patients, all randomized Stage II-IIIA patients and Intent to Treat (ITT) Stage IB-IIIA populations. Key Secondary endpoints included Overall Survival (OS) in the overall study population and ITT Stage IB-IIIA NSCLC patients. At data cutoff on January 21, 2021, median follow up was 32.2 months in the ITT population.
Treatment with TECENTRIQ® following surgery and chemotherapy reduced the risk of disease recurrence or death (DFS) by 34% (HR=0.66; P=0.0039), in patients with Stage II-IIIA NSCLC, whose tumor PD-L1 expression was 1% or more, compared with BSC. In this patient population, median DFS was Not Reached for TECENTRIQ®, compared with 35.3 months for BSC.
In the larger population of all randomized Stage II-IIIA study patients, TECENTRIQ® reduced the risk of disease recurrence or death by 21% (HR=0.79, P=0.02). In this patient population, TECENTRIQ® increased DFS by a median of seven months, compared with BSC (42.3 months versus 35.3 months).
The significance boundary was not crossed for DFS in the ITT patient population. Overall Survival data were immature and not formally tested. Safety data for TECENTRIQ® were consistent with its known safety profile and no new safety signals were identified.
It was concluded that this study met its Primary endpoint, and is the first Phase III study to demonstrate that treatment with TECENTRIQ® following surgery and chemotherapy can significantly delay disease recurrence in patients with early stage lung cancer, with a more pronounced benefit noted, in patients with tumor PD-LI expression of 1% or more.
IMpower010: Primary results of a phase III global study of atezolizumab versus best supportive care after adjuvant chemotherapy in resected stage IB-IIIA non-small cell lung cancer (NSCLC). Wakelee HA, Altorki NK, Zhou C, et al. J Clin Oncol. 2021;39:(suppl 15; abstr 8500). doi:10.1200/JCO.2021.39.15_suppl.8500
FDA Approves LUMAKRAS® for KRAS G12C-Mutated Non Small Cell Lung Cancer
SUMMARY: The FDA on May 28, 2021, granted accelerated approval to LUMAKRAS® (Sotorasib), a RAS GTPase family inhibitor, for adult patients with KRAS G12C mutated locally advanced or metastatic Non Small Cell Lung Cancer (NSCLC), as determined by an FDA approved test, who have received at least one prior systemic therapy. The FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for LUMAKRAS®. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The KRAS (kirsten rat sarcoma viral oncogene homologue) proto-oncogene encodes a protein that is a member of the small GTPase super family. The KRAS gene provides instructions for making the KRAS protein, which is a part of a signaling pathway known as the RAS/MAPK pathway. By relaying signals from outside the cell to the cell nucleus, the protein instructs the cell to grow, divide and differentiate. The KRAS protein is a GTPase, and converts GTP into GDP. To transmit signals, the KRAS protein must be turned on, by binding to a molecule of GTP. When GTP is converted to GDP, the KRAS protein is turned off or inactivated, and when the KRAS protein is bound to GDP, it does not relay signals to the cell nucleus. The KRAS gene is in the Ras family of oncogenes, which also includes two other genes, HRAS and NRAS. When mutated, oncogenes have the potential to change normal cells cancerous.
KRAS is the most frequently mutated oncogene in human cancers and are often associated with resistance to targeted therapies and poor outcomes. The KRAS-G12C mutation occurs in approximately 12-15% of Non Small Cell Lung Cancers (NSCLC) and in 3-5% of Colorectal cancers and other solid cancers. KRAS G12C is one of the most prevalent driver mutations in NSCLC and accounts for a greater number of patients than those with ALK, ROS1, RET, and TRK 1/2/3 mutations combined. KRAS G12C cancers are genomically more heterogeneous and occur more frequently in current or former smokers, and are likely to be more complex genomically than EGFR mutant or ALK rearranged cancers. G12C is a single point mutation with a Glycine-to-Cysteine substitution at codon 12. This substitution favors the activated state of KRAS, resulting in a predominantly GTP-bound KRAS oncoprotein, amplifying signaling pathways that lead to oncogenesis.
LUMAKRAS® is a first-in-class small molecule that specifically and irreversibly inhibits KRAS-G12C and traps KRAS-G12C in the inactive GDP-bound state. Preclinical studies in animal models showed that LUMAKRAS® inhibited nearly all detectable phosphorylation of Extracellular signal-Regulated Kinase (ERK), a key downstream effector of KRAS, leading to durable complete regression of KRAS-G12C tumors.
The CodeBreaK clinical development program for LUMAKRAS® was designed to treat patients with an advanced solid tumor with the KRAS G12C mutation and address the longstanding unmet medical need for these cancers. This program has enrolled more than 800 patients across 13 tumor types since its inception.
CodeBreaK 100 is a Phase I and II, first-in-human, open-label, single arm, multicenter study, which enrolled patients with KRAS G12C-mutant solid tumors. Eligible patients must have received a prior line of systemic anticancer therapy, for their tumor type and stage of disease. The present FDA approval was based on a Phase II trial which enrolled 126 patients with NSCLC, 124 of whom had centrally evaluable lesions by RECIST criteria at baseline. Enrolled patients had locally advanced or metastatic NSCLC with a KRAS G12C mutation, who had progressed on an immune checkpoint inhibitor and/or platinum-based chemotherapy, and those with active brain metastases were excluded. Patient received LUMAKRAS® 960mg orally once daily, until disease progression or unacceptable toxicity. Imaging studies were done every 6 weeks up to week 48 and then once every 12 weeks thereafter. The Primary end point of the trial was Overall Response Rate (ORR) as assessed by blinded Independent Central Review. Secondary end points included Duration of Response (DOR), Disease Control Rate (DCR), time to recovery, Progression Free Survival (PFS), Overall Survival, and Safety. The examination of biomarkers served as an exploratory end point. Patients were followed for a median of 12.2 months.
The ORR was 37.1% and the median Duration of Response was 10 months. Three patients had a Complete Response and the Disease Control Rate was 80.6%. The median Time to response was 1.4 months and 72% of patients had an early rapid response on first CT scan at 6 weeks. Approximately 81% of patients had tumor shrinkage of any magnitude, and the median percentage of best tumor shrinkage among all responders was 60%, and these responses were durable. The median PFS was 6.8 months. In the exploratory biomarker analysis, tumor response to LUMAKRAS® was seen across subgroups, including patients with negative or low expression of PD-L1 and those with STK11 and TP53 mutations. The most common adverse reactions were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities were increase in liver function tests, anemia, hyponatremia and proteinuria.
It was concluded that patients with NSCLC have poor outcomes and limited treatment options following progression on first line treatment. LUMAKRAS® offers a new treatment option for this patient group, and it is the first KRAS-targeted therapy to be approved after nearly four decades of research. A global Phase III study (CodeBreaK 200) is underway, comparing LUMAKRAS® to Docetaxel in patients with KRAS G12C-mutated NSCLC.
CodeBreaK 100: Registrational Phase 2 Trial of Sotorasib in KRAS p.G12C Mutated Non-small Cell Lung Cancer. Li BT, Skoulidis F, Falchook G, et al. Presented at: International Association for the Study of Lung Cancer 2020 World Conference on Lung Cancer; January 28-31, 2021; virtual. Abstract PS01.07.
FDA Approves Bispecific Antibody RYBREVANT® for Metastatic Non Small Cell Lung Cancer
SUMMARY: The FDA on May 21, 2021, granted accelerated approval to RYBREVANT® (Amivantamab-vmjw), a bispecific antibody directed against Epidermal Growth Factor (EGF) and MET receptors, for adult patients with locally advanced or metastatic Non Small Cell Lung Cancer (NSCLC) with Epidermal Growth Factor Receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after Platinum-based chemotherapy. FDA also approved the Guardant360® CDx (Guardant Health, Inc.) as a companion diagnostic for RYBREVANT®.
The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR mutations and 90% of these mutations are either exon 19 deletions or L858R substitution mutation in exon 21. EGFR exon 20 insertion mutations are the third most common after L858R and exon 19 deletions, and occur in about 2-3% patients with NSCLC and are insensitive to EGFR Tyrosine Kinase Inhibitors (TKIs) due to an altered conformation of the kinase active site. Next-Generation sequencing provides an alternative to Polymerase Chain Reaction (PCR)-based tests, which fail to identify 50% or more of exon 20 insertion mutations. Patients with EGFR exon 20 insertion mutations have a 5 year Overall Survival (OS) of 8% in the frontline setting, compared to an OS of 19% for patients with EGFR exon 19 deletions or L858R mutations. There is therefore a clinically unmet need for this patient group, as there are no approved targeted therapies available and platinum-doublet chemotherapy remains the standard of care for these patients.
Epidermal Growth Factor Receptor (EGFR) plays an important role in regulating cell proliferation, survival and differentiation, and is overexpressed in a variety of epithelial malignancies. EGFR targeted Tyrosine Kinase Inhibitors (TKIs) such as Gefitinib, Erlotinib, Afatinib, Dacomitinib and Osimertinib target the EGFR signaling cascade. However, patients eventually will develop drug resistance due to new EGFR mutations. Another important cause of drug resistance to TKIs is due to the activation of parallel RTK (Receptor Tyrosine Kinase) pathways such as Hepatocyte Growth Factor/Mesenchymal-Epithelial Transition factor (HGF/MET) pathway, thereby bypassing EGFR TKI inhibitors.
RYBREVANT® is a fully-human bispecific antibody directed against EGFR and MET receptors. RYBREVANT® binds extracellularly and simultaneously blocks ligand-induced phosphorylation of EGFR and c-MET, inhibiting tumor growth and promoting tumor cell death. Further, RYBREVANT® downregulates receptor expression on tumor cells thus preventing drug resistance mediated by new emerging mutations of EGFR or c-MET. By binding to the extracellular domain of the receptor protein, RYBREVANT® can bypass primary and secondary TKI resistance at the active site.
The present FDA approval was based on CHRYSALIS, an ongoing multicenter, non-randomized, open label, multicohort, Phase I clinical trial (NCT02609776) which included patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. The purpose of study is to evaluate the safety, pharmacokinetics, and preliminary efficacy of RYBREVANT® as a monotherapy and in combination with Lazertinib, and to determine the recommended Phase 2 dose (RP2D) (monotherapy), recommended Phase 2 combination dose (RP2CD) (combination therapy), and to determine recommended Phase 2 Dose (RP2q3W) with combination chemotherapy (RYBREVANT® in combination with standard of care Carboplatin and Pemetrexed) in 21 day treatment cycle for participants with advanced NSCLC.
In this analysis of the Phase 1 CHRYSALIS study, researchers assessed the efficacy and safety of RYBREVANT® in patients with NSCLC and EGFR exon 20 insertion mutations, who had progressed on prior Platinum-based chemotherapy, and were treated at the recommended Phase II dose of RYBREVANT® 1050 mg (1400 mg for patients weighting 80 kg or more). The median patient age was 61 years, 51% were female, and median prior lines of therapy was one. The Primary endpoint was Overall Response Rate (ORR). Secondary endpoints included Duration of Response (DOR), Clinical Benefit Rate, Progression Free Survival (PFS) and Overall Survival (OS).
It was noted that among this post-platinum cohort of patients (N=81), at a median follow up of 9.7 months, the ORR was 40%, with 4% Complete Reponses and 36% achieving Partial Responses (PR). Responses were durable with median Duration of Response of 11.1 months, with 63 % having responses of at least six months or greater duration. The median PFS was 8.3 months and median OS was 22.8 months. The Clinical Benefit Rate (PR or more, or Stable Disease of 11 weeks or more) was 74%. The most common adverse reactions (20% or more) were rash, infusion-related reactions, paronychia, fatigue, musculoskeletal pain, stomatitis, nausea, vomiting, constipation, edema, cough and dyspnea.
The authors concluded that RYBREVANT® demonstrated robust and durable antitumor activity in patients with EGFR exon 20 insertion mutations, with a manageable safety profile.
Amivantamab in Post-platinum EGFR Exon 20 Insertion Mutant Non-small Cell Lung Cancer. Sabari JK, Shu CA, Park K, et al. Presented at: IASLC 2020 World Conference on Lung Cancer Singapore. January 28-31, 2021. Abstract OA04.04
Five-Year Efficacy Outcomes with KEYTRUDA® versus Chemotherapy in Metastatic NSCLC
SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas.
Immunotherapy with Immune Checkpoint Inhibitors (ICIs) has revolutionized cancer care and has become one of the most effective treatment options, by improving Overall Response Rate and prolongation of survival across multiple tumor types. Immune Checkpoint Inhibitors (ICIs) target Programmed cell Death protein-1 (PD-1) receptors on T cells, as well as Programmed cell Death Ligand-1 (PD-L1), PD-L2 and Cytotoxic T-Lymphocyte-Associated protein-4 (CTLA-4), and many other important regulators of the immune system, which are upregulated in some tumor types. T-cell proliferation and cytokine production is inhibited upon binding of the PD-1 ligands PD-L1 and PD-L2, to the PD-1 receptor found on T cells.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, releasing PD-1 pathway-mediated inhibition of the immune response. Unleashing the T cells results in T cell proliferation, activation and a therapeutic response. High level of PD-L1 expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression, and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.
KEYNOTE-024 is an open-label, randomized, Phase III trial in which KEYTRUDA® administered at a fixed dose was compared with investigator’s choice of cytotoxic chemotherapy, as first line therapy, for patients with advanced NSCLC, with tumor PD-L1 expression of 50% or greater. Three hundred and five (N=305) treatment naïve patients with advanced NSCLC and PD-L1 expression on at least 50% of tumor cells, were randomly assigned in a 1:1 ratio to receive either KEYTRUDA® (N=154) or chemotherapy (N=151). Enrolled patients had no sensitizing EGFR mutations or ALK translocations. Treatment consisted of KEYTRUDA® administered at a fixed dose of 200 mg IV every 3 weeks for up to 2 years or the investigator’s choice of Platinum-based chemotherapy for 4-6 cycles. Pemetrexed (ALIMTA®) based therapy was permitted only for patients who had non-squamous tumors and these patients could receive ALIMTA® maintenance therapy after the completion of combination chemotherapy. Patients in the chemotherapy group who experienced disease progression were allowed to cross over to the KEYTRUDA® group. The Primary end point was Progression Free Survival (PFS) and Secondary end points included Overall Survival (OS), Objective Response Rate (ORR) and Safety. In an updated analysis of the KEYNOTE-024 study, after a median follow up of 25.2 months, the median OS was 30 months in the KEYTRUDA® group and 14.2 months in the chemotherapy group (HR=0.63; P=0.002). This OS benefit was maintained even after adjusting for crossover.
The authors in this publication reported the 5-year efficacy and safety outcomes from this pivotal Phase III KEYNOTE-024 trial. The median time from randomization to data cutoff was 59.9 months. Among patients initially assigned to chemotherapy, 66% received subsequent anti PD-1 or PD-L1 therapy (66% cross over rate). In the KEYTRUDA® group, 52.9% received additional anticancer therapy.
The median OS was 26.3 months for KEYTRUDA® and 13.4 months for chemotherapy (HR=0.62). Kaplan-Meier estimates of the 5-year OS rate were 31.9% for the KEYTRUDA group and 16.3% for the chemotherapy group. The ORR was 46.1% among patients in the KEYTRUDA® group versus 31.1% in the chemotherapy group and the median Duration of Response was 29.1 months in the KEYTRUDA® group and 6.3 months in the chemotherapy group.
The authors concluded that first line KEYTRUDA® provides a durable and clinically meaningful long-term Overall Survival benefit, when compared to chemotherapy, in patients with metastatic NSCLC, with PD-L1 Tumor Proportion Score of at least 50%.They added that this is first 5-year follow up of any first line Phase III immunotherapy trial for Non Small Cell Lung Cancer.
Five-Year Efficacy Outcomes With Pembrolizumab vs Chemotherapy in Metastatic NSCLC With PD-L1 Tumor Proportion Score of at Least 50%: KEYNOTE-024 Trial. Reck M , Rodríguez–Abreu D, Robinson AG, et al. DOI: 10.1200/JCO.21.00174 Journal of Clinical Oncology. Published online April 19, 2021.
FDA Approves LIBTAYO® for Non Small Cell Lung Cancer with High PD-L1 Expression
SUMMARY: The FDA on February 22, 2021, approved LIBTAYO® (Cemiplimab-rwlc) for the first line treatment of patients with advanced Non Small Cell Lung Cancer (NSCLC) (locally advanced who are not candidates for surgical resection or definitive chemoradiation or metastatic), whose tumors have high PD-L1 expression (Tumor Proportion Score [TPS] 50% or more), as determined by an FDA-approved test, with no EGFR, ALK or ROS1 aberrations.
The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Immunotherapy with Immune Checkpoint Inhibitors (ICIs) has revolutionized cancer care and has become one of the most effective treatment options, by improving Overall Response Rate and prolongation of survival, across multiple tumor types.
Available Immune Checkpoint Inhibitors (ICIs) target Programmed cell Death protein-1 (PD-1) receptors on T cells, as well as Programmed cell Death Ligand-1 (PD-L1), PD-L2 and Cytotoxic T-Lymphocyte-Associated protein-4 (CTLA-4), and many other important regulators of the immune system, which are upregulated in some tumor types. T-cell proliferation and cytokine production is inhibited upon binding of the PD-1 ligands PD-L1 and PD-L2, to the PD-1 receptor found on T cells.
LIBTAYO® is a recombinant human immunoglobulin G4 (IgG4) monoclonal antibody that binds to PD-1 and blocks its interaction with PD-L1 and PD-L2, releasing PD-1 pathway-mediated inhibition of the immune response. Unleashing the T cells results in T cell proliferation, activation and a therapeutic response. LIBTAYO® is indicated for the treatment of subsets of patients with advanced Basal Cell Carcinoma and advanced cutaneous Squamous Cell Carcinoma.
The present FDA approval of LIBTAYO® is based on EMPOWER-Lung 1, which is a multicentre, open-label, global, Phase III trial, which examined the benefit of LIBTAYO® in the first-line treatment of advanced NSCLC with PD-L1 expression of at least 50%. In this study, 710 (N=710) patients (intent-to-treat) with Squamous or non-Squamous, locally advanced NSCLC who were not candidates for surgical resection or definitive chemoradiation, or with metastatic NSCLC were randomized (1:1) to receive LIBTAYO® 350 mg IV every 3 weeks for up to 108 weeks (N=356) or 4-6 cycles of investigator’s choice of platinum doublet chemotherapy (N=354). The most common chemotherapy regimens selected were Carboplatin plus Paclitaxel, Carboplatin plus Pemetrexed, and Carboplatin plus Gemcitabine. Crossover from chemotherapy to LIBTAYO® was allowed following disease progression, and never-smokers were not eligible for the trial. The co-Primary end points of the study were Overall Survival (OS) and Progression Free Survival (PFS), per the Blinded Independent Review Committee. Primary endpoints were assessed in the intention-to-treat population and in a prespecified population of patients with PD-L1 of at least 50%. Secondary end points included Overall Response Rate (ORR), Duration of Response (DOR), Health-Related Quality of Life (HRQoL), and Safety.
This trial demonstrated statistically significant improvements in OS and PFS for patients receiving LIBTAYO® compared to those treated with platinum-based chemotherapy, despite a high crossover rate (74%). The median OS was 22.1 months with LIBTAYO® versus 14.3 months with chemotherapy (HR=0.68; P=0.0022), demonstrating that LIBTAYO® reduced the risk of death by 32% compared to chemotherapy. An additional analysis of 563 patients with proven PD-L1 expression of 50% or higher found that the median OS was Not Reached with LIBTAYO® (N=283) versus 14.2 months with chemotherapy (N=280). LIBTAYO® reduced the risk of death by 43% compared to chemotherapy HR=0.57; P=0.0002). The median PFS was 6.2 months in the LIBTAYO® group and 5.6 months in the chemotherapy group (HR= 0.59; P<0.0001). Among those with PD-L1 expression of 50% or higher, the median PFS was 8.2 months with LIBTAYO® versus 5•7 months with chemotherapy (HR=0•54; P<0•0001). The confirmed ORR was 37% and 21% in the LIBTAYO® and chemotherapy arms respectively, and the median DOR was 21.0 months in the LIBTAYO® arm versus 6.0 months in the chemotherapy arm.
The authors concluded that LIBTAYO® monotherapy significantly improved Overall Survival and Progression Free Survival compared with chemotherapy, in patients with advanced Non Small Cell Lung Cancer with PD-L1 of at least 50%, providing a potential new treatment option for this patient population.
Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Sezer A, Kilickap S, Gümüş M, et al. Lancet. 2021;397:592-604. doi: 10.1016/S0140-6736(21)00228-2.
Therapy for Stage IV Non–Small-Cell Lung Cancer with Driver Alterations: ASCO and OH (CCO) Joint Guideline Update
SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8%, as well as other mutations in BRAF, MET, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive, and the presence of two simultaneous mutations, are rare.
The ASCO and Ontario Health (Cancer Care Ontario) NSCLC Expert Panel updated the 2017 ASCO guideline on systemic therapy for patients with Stage IV NSCLC with driver alterations and provided evidence-based recommendations, based on a systematic review of Randomized Controlled Trials (RCTs) from December 2015 to January 2020 and meeting abstracts from ASCO 2020.
This clinical practice guideline addresses three comprehensive clinical questions for patients with Stage IV NSCLC with driver alterations
1) What is the most effective first-line therapy?
2) What is the most effective second-line therapy?
3) Is there a role for a third-line therapy or beyond?
The guideline addresses patients with NSCLC in the following histologic or subgroups: EGFR, ALK, ROS1, BRAF, MET, RET, HER2, and NTRK. This update does not apply to patients with Stage IV NSCLC without known driver alterations and those with rarer histologies such as large cell, neuroendocrine, etc.
Summary of Key Recommendations
Recommendation 1.1: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the first-line setting, for patients with T790M, L858R, or exon 19 deletion mutations, Osimertinib should be offered.
Recommendations 1.2, 1.3, 1.4, and 1.5: For patients with Stage IV NSCLC and driver alterations in EGFR-if Osimertinib is not available
֍In the first-line setting, if Osimertinib is not available, Gefitinib with chemotherapy may be offered or Dacomitinib may be offered.
֍Other options that may be offered include Afatinib or Erlotinib/Bevacizumab or Erlotinib/Ramucirumab or Gefitinib, Erlotinib, or Icotinib.
Recommendation 1.6: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the first-line setting, for patients with a Performance Status (PS) of 3, an EGFR Tyrosine Kinase Inhibitor (TKI) may be offered.
Recommendation 1.7: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the first-line setting, for patients with EGFR mutations other than exon 20 insertion mutations, T790M, L858R, or exon 19 deletion alterations, Afatinib may be offered or Osimertinib may be offered or treatments outlined in the ASCO/OH nondriver mutation guideline may be offered.
Recommendation 1.8: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the first-line setting, for patients with any activating EGFR mutation (including exon 20 insertion mutations), regardless of Programmed Death Ligand-1 (PD-L1) expression levels, single-agent immunotherapy should not be used.
Recommendation 1.9: For patients with Stage IV NSCLC and driver alterations in EGFR causing resistance to first- and second-generation EGFR TKIs
֍In the first-line setting, for patients with EGFR exon 20 insertion mutation causing resistance to first- and second-generation EGFR TKIs, doublet chemotherapy with or without Bevacizumab or standard treatment outlined in the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 2.1 and 2.2: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the second-line setting, for patients who did not receive Osimertinib and have a T790M mutation at the time of progressive disease, Osimertinib should be offered.
֍In the second-line setting, for patients with any EGFR mutation who have progressed on EGFR TKIs with no T790M mutation OR whose disease has progressed on Osimertinib, treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendation 3.1: For patients with Stage IV NSCLC and driver alterations in ALK
֍In the first-line setting, Alectinib or Brigatinib should be offered.
֍In the first-line setting, if Alectinib and Brigatinib are not available, Ceritinib or Crizotinib should be offered.
Recommendations 4.1, 4.2, and 4.3: For patients with stage IV NSCLC and driver alterations in ALK
֍In the second-line setting, if Alectinib or Brigatinib was given in the first-line setting, Lorlatinib may be offered.
֍In the second-line setting, if Crizotinib was given in the first-line setting, then Alectinib, Brigatinib, or Ceritinib should be offered.
֍In the third-line setting, if Crizotinib was given in the first-line setting and Alectinib, Brigatinib, or Ceritinib in the second-line setting, then Lorlatinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 5.1, 5.2, and 5.3: For patients with Stage IV NSCLC and driver alterations in ROS1
֍In the first-line setting, Crizotinib or Entrectinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered or Ceritinib or Lorlatinib may be offered.
Recommendations 6.1 and 6.2: For patients with Stage IV NSCLC and driver alterations in ROS1
֍In the second-line setting, if ROS1-targeted therapy was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
֍In the second-line setting, if nontargeted therapy was given in the first-line setting, Crizotinib, Ceritinib, or Entrectinib may be offered.
Recommendations 7.1 and 7.2: For patients with Stage IV NSCLC and driver alterations with BRAF V600E mutation
֍In the first-line setting, Dabrafenib/Trametinib may be offered or standard first-line treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 8.1, 8.2 and 8.3: For patients with Stage IV NSCLC and driver alterations with BRAF V600E mutation
֍In the second-line setting, if previous BRAF/MEK-targeted therapy (Dabrafenib/Trametinib) was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
֍In the second-line setting, if BRAF-targeted therapy was not given in the first-line setting, Dabrafenib/Trametinib may be offered or Dabrafenib or Vemurafenib alone may be offered.
֍If previous chemotherapy, immunotherapy, and/or BRAF-targeted therapy were given in the first- or subsequent-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
Recommendation 8.4: For patients with Stage IV NSCLC and driver alterations with BRAF mutations other than V600E
֍In the second-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
Recommendations 9.1 and 9.2: For patients with Stage IV NSCLC and MET exon 14 skipping mutation
֍In the first-line setting, for patients with an MET exon 14 skipping mutation, MET-targeted therapy with Capmatinib or Tepotinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 10.1 and 10.2: For patients with Stage IV NSCLC and MET exon 14 skipping mutation
֍In the second-line setting, for MET abnormalities other than exon 14 skipping mutations or if MET-targeted therapy was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
֍In the second-line setting, patients with an MET exon 14 skipping mutation who previously received or were ineligible for first-line chemotherapy with or without immunotherapy (ie. if MET-targeted therapy was not given in the first-line setting), Capmatinib or Tepotinib may be offered.
Recommendations 11.1, 11.2, and 11.3: For patients with Stage IV NSCLC and driver alterations in RET
֍In the first-line setting, Selpercatinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered or Pralsetinib may be offered.
Recommendations 12.1, 12.2, and 12.3: For patients with Stage IV NSCLC and driver alterations in RET
֍In the second-line setting, if RET-targeted therapy was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
֍In the second-line setting, if RET-targeted therapy was not given in the first-line setting, Selpercatinib may be offered or Pralsetinib may be offered.
Recommendations 13.1 and 13.2: For patients with Stage IV NSCLC and driver alterations in NTRK
֍In the first-line setting, Entrectinib or Larotrectinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 14.1 and 14.2: For patients with Stage IV NSCLC and driver alterations in NTRK
֍In the second-line setting, if NTRK-targeted therapy was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
֍In the second-line setting, if NTRK-targeted therapy was not given in the first-line setting, Entrectinib or Larotrectinib may be offered.
Therapy for Stage IV Non–Small-Cell Lung Cancer With Driver Alterations: ASCO and OH (CCO) Joint Guideline Update. Hanna NH, Robinson AG, Temin S, et al. J Clin Oncol. 2021;39: 1040-1091
FDA Approves LORBRENA® for Advanced ALK-Positive Lung Cancer
SUMMARY: The FDA on March 3, 2021, granted regular approval to LORBRENA® (Lorlatinib) for patients with metastatic Non Small Cell Lung Cancer (NSCLC) whose tumors are Anaplastic Lymphoma Kinase (ALK)-positive, as detected by an FDA-approved test. The FDA also approved the Ventana ALK (D5F3) CDx Assay (Ventana Medical Systems, Inc.) as a companion diagnostic for LORBRENA®. Lung cancer is the leading cause of cancer death in both men and women, and accounts for about 14% of all new cancers and 25% of all cancer deaths. The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The discovery of chromosomal rearrangements of the Anaplastic Lymphoma Kinase (ALK) gene in some patients with advanced NSCLC and adenocarcinoma histology, and their sensitivity to ALK inhibitors, paved the way to the development of small-molecule ALK Tyrosine Kinase Inhibitors. It has become clear that appropriate, molecularly targeted therapy for tumors with a molecular abnormality, results in the best outcomes. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8%, as well as other mutations in BRAF, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive, and the presence of two simultaneous mutations, are rare.
ALK inhibitors include first-generation XALKORI® (Crizotinib) and second-generation ALK inhibitors such as ZYKADIA® (Ceritinib), ALECENSA® (Alectinib) and ALUNBRIG® (Brigatinib). Despite the improved efficacy of second-generation ALK inhibitors, recurrent disease due to drug resistance including CNS disease progression, can still develop.
LORBRENA® is a novel third-generation ALK inhibitor that is more potent than second-generation inhibitors, and has the broadest coverage of ALK resistance mutations that have been identified. LORBRENA® crosses the blood-brain barrier and has marked intracranial activity in previously treated patients with baseline CNS disease, including leptomeningeal disease. LORBRENA® received accelerated approval by the FDA in November 2018 for the second or third-line treatment of ALK-positive metastatic NSCLC. However, the efficacy of LORBRENA®, as compared with that of XALKORI®, as first line treatment for advanced ALK-positive NSCLC, has been unclear.
The CROWN trial is a global, open label, randomized, Phase 3 study, in which LORBRENA® was compared with XALKORI®, in patients with previously untreated ALK-positive advanced NSCLC. In this study, 296 treatment naïve advanced NSCLC patients were randomly assigned 1:1 to receive LORBRENA® 100 mg orally once daily (N=149) or XALKORI® 250 mg orally twice daily (N=147) in cycles of 28 days. Treatment was continued until disease progression or unacceptable toxic effects. Eligible patients were required to have ALK-positive tumors detected by the Ventana ALK (D5F3) CDx assay. Patients with asymptomatic treated or untreated CNS metastases were eligible and had to have at least one extracranial measurable target lesion that had not been previously irradiated. Patients were stratified according to the presence of brain metastases and ethnic group (Asian or non-Asian) and crossover between the treatment groups was not permitted. The Primary end point was Progression Free Survival (PFS) as assessed by Blinded Independent Central Review (BICR). Secondary end points included independently assessed Objective Response Rate (ORR) and intracranial response.
At a planned interim analysis, treatment with LORBRENA® resulted in statistically significant and clinically meaningful improvement in PFS as assessed by BICR, with a Hazard Ratio of 0.28 (P<0.001), corresponding to a 72% reduction in the risk of disease progression or death. The median PFS was not estimable in the LORBRENA® arm and was 9.3 months for those treated with XALKORI®. The percentage of patients who were alive without disease progression at 12 months was 78% in the LORBRENA® group and 39% in the XALKORI® group, and the Hazard Ratio favored LORBRENA® over XALKORI® across all prespecified patient subgroups. The Overall Survival data were immature at the PFS analysis.
The confirmed ORR was 76% with LORBRENA® and 58% with XALKORI®. About 70% of the patients who received LORBRENA® and 27% of those who received XALKORI® had a response that lasted at least 12 months. Additionally, treatment with LORBRENA® was associated with increased intracranial activity compared with XALKORI®. Among patients presenting with measurable brain metastases, the intracranial ORR was 82% with LORBRENA® and 23% with XALKORI®, with a intracranial Complete Response rate of 71% and 8%, respectively. The duration of intracranial response was 12 months or more in 79% and 0% of patients in the LORBRENA® and XALKORI® groups, respectively. The most common adverse events with LORBRENA® were hyperlipidemia, edema, weight gain, peripheral neuropathy, and cognitive effects.
It was concluded that treatment LORBRENA® resulted in a significantly longer Progression Free Survival and a higher frequency of intracranial response, compared to XALKORI®, among patients with previously untreated advanced ALK-positive NSCLC.
First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. Shaw AT, Bauer TM, de Marinis F, et al. N Engl J Med 2020; 383:2018-2029.
Advances with First-Line Dual Immunotherapies in Metastatic Non-Small Cell Lung Cancer
By Dr. David Waterhouse | Sponsored by Bristol Myers Squibb
Dr. Waterhouse is a paid consultant for Bristol Myers Squibb and was compensated for his role in drafting this article.
The American Cancer Society estimates that there will be nearly 229,000 new cases of lung cancer in the United States (US) alone in 2020 and nearly 136,000 lung cancer deaths.1 Historically, most patients present with metastatic disease and their long-term outlook is grim.2 However, significant progress has been made in recent years. In August 2020, Howlader et al reported that the population-level mortality from non-small cell lung cancer (NSCLC) in the US fell sharply from 2013 to 2016.3
Based on the results from Checkmate 227 Part 1a, OPDIVO, in combination with YERVOY, is indicated for the first-line treatment of adult patients with metastatic NSCLC whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.4-6 In addition, based on the results from Checkmate 9LA, OPDIVO, in combination with YERVOY and 2 cycles of platinum-doublet chemotherapy (chemo), is indicated for the first-line treatment of adult patients with metastatic or recurrent NSCLC, with no EGFR or ALK genomic tumor aberrations.4,6,7
OPDIVO and YERVOY are associated with the following Warnings and Precautions: severe and fatal immune-mediated reactions including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis with renal dysfunction, dermatologic adverse reactions, other immune-mediated adverse reactions; infusion-related reactions; complications of allogeneic hematopoietic stem cell transplantation (HSCT); embryo-fetal toxicity; and increased mortality in patients with multiple myeloma when OPDIVO is added to a thalidomide analogue and dexamethasone, which is not recommended outside of controlled clinical trials.4 Please see additional Important Safety Information for OPDIVO and YERVOY at the end of the article and US Full Prescribing Information for OPDIVO and YERVOY at https://packageinserts.bms.com/pi/pi_opdivo.pdf and https://packageinserts.bms.com/pi/pi_yervoy.pdf.
OPDIVO® (nivolumab) is a monoclonal antibody targeting programmed death receptor-1 (PD-1) that has been approved for the treatment of lung cancer.4 YERVOY® (ipilimumab) is another monoclonal antibody that works to activate the immune system by targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4).6,8
The phase 3 Checkmate 227 and Checkmate 9LA trials investigated OPDIVO plus YERVOY-based combinations for first-line treatment of certain NSCLC patients.4 Part 1a of Checkmate 227 investigated the effects of OPDIVO + YERVOY compared with standard chemo* among patients whose tumors expressed ≥1% programmed death ligand 1 (PD-L1)4
OPDIVO + YERVOY showed a superior survival benefit compared with chemo*, with the primary analysis at a minimum follow-up of 29.3 months revealing a median overall survival (OS) of 17.1 months vs 14.9 months with chemo*, and a hazard ratio (HR) of 0.79, 95% confidence interval (CI): 0.67–0.94, P=0.0066 (Figure 3).4,16 The median progression-free survival (PFS) was 5.1 months (95% CI: 4.1–6.3) with OPDIVO + YERVOY and 5.6 months (95% CI: 4.6–5.8) with chemo* alone (HR=0.82; 95% CI: 0.69–0.97).4
The most frequent (≥2%) serious adverse reactions were pneumonia, diarrhea/colitis, pneumonitis, hepatitis, pulmonary embolism, adrenal insufficiency, and hypophysitis. Fatal adverse reactions occurred in 1.7% of patients; these included events of pneumonitis (4 patients), myocarditis, acute kidney injury, shock, hyperglycemia, multi-system organ failure, and renal failure.4 The most common (≥20%) adverse reactions were fatigue (44%), rash (34%), decreased appetite (31%), musculoskeletal pain (27%), diarrhea/colitis (26%), dyspnea (26%), cough (23%), hepatitis (21%), nausea (21%), and pruritus (21%).4 Please continue reading for more Important Safety Information for OPDIVO and YERVOY throughout.
At the American Society for Clinical Oncology (ASCO) 2020 Annual Meeting, 3-year follow-up results from this trial were reported. With a median follow-up of more than 3 years (43.1 months), this study represents the longest median follow-up of any dual immuno-oncology (I-O)-based combination in a phase 3 clinical trial in NSCLC.15 This extended follow-up analysis showed 3-year OS rates of 33% for OPDIVO + YERVOY and 22% for chemo*
At minimum follow-up of 28.3 months, the objective response rate was 36% (95% CI: 31–41), CR=5.8%, PR=30.1% with OPDIVO + YERVOY and 30% (95% CI: 26–35), CR=1.8%, PR=28.2% with chemo*.4,16,17 The median duration of response from the extended 3-year follow-up analysis was 23.2 months (95% CI: 15.2–32.2) in patients who responded to OPDIVO + YERVOY and 6.7 months (95% CI: 5.6–7.6) with chemo*
The 3-year data from Checkmate 227 Part 1a show the long-term durable survival of a dual immunotherapy approach.15 The FDA approved OPDIVO + YERVOY on May 15, 2020, for first-line treatment of adult patients with metastatic NSCLC whose tumors express PD-L1(≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations. With this approval, these patients with NSCLC can now be offered the option of dual I-O therapy.4,5
Also reported at ASCO 2020 were the results of Checkmate 9LA.18 Patients were randomized to receive either the combination of OPDIVO + YERVOY and 2 cycles of platinum-doublet chemo† or platinum-doublet chemo† for 4 cycles.4 This trial evaluated patients regardless of PD-L1 expression and histology
The trial showed a superior benefit in OS for patients treated with OPDIVO + YERVOY with limited chemo† compared to those who received chemo† alone.18 At the pre-specified interim analysis at 8.1 months, the median OS was 14.1 months vs 10.7 months (HR=0.69, 96.71% CI: 0.55-0.87, P=0.0006).4 Median PFS per blinded independent central review (BICR) at minimum follow-up of 6.5 months was 6.8 months among patients who received OPDIVO + YERVOY with chemo†, and 5.0 months among patients receiving chemo† (HR=0.70, 97.48% CI: 0.57-0.86).4 Confirmed ORR per BICR at minimum follow-up of 6.5 months was 38% (95% CI: 33-43) and 25% (95% CI: 21-30) respectively.4,18
A follow-up analysis performed at 12.7 months showed median OS of 15.6 months with OPDIVO + YERVOY with chemo† and 10.9 months with chemo† alone with HR of 0.66 (95% CI: 0.55-0.80) (Figure 6).4,18 OS was consistent across PD-L1 expression levels at minimum follow-up of 8.1 months, with median OS of 14.0 months (95% CI:13.2-NR) and 10.0 months (95% CI: 7.7-13.7) in patients treated with OPDIVO + YERVOY with limited chemo† and chemo† respectively in the PD-L1 <1% sub-population (HR=0.65), and median OS of 14.2 months (95% CI:13.1-NR) and 10.6 months (95% CI: 9.4-12.6) respectively (HR=0.67) in the PD-L1 ≥1% sub-population.19
In this study, the most frequent (>2%) serious adverse reactions were pneumonia, diarrhea, febrile neutropenia, anemia, acute kidney injury, musculoskeletal pain, dyspnea, pneumonitis, and respiratory failure. Fatal adverse reactions occurred in 7 (2%) patients, and included hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.4 The most common (>20%) adverse reactions were fatigue (49%), musculoskeletal pain (39%), nausea (32%), diarrhea (31%), rash (30%), decreased appetite (28%), constipation (21%), and pruritus (21%).4 Please continue reading for more Important Safety Information for OPDIVO and YERVOY throughout. The FDA approved OPDIVO, in combination with YERVOY and 2 cycles of platinum-doublet chemo, for the first-line treatment of adult patients with metastatic or recurrent NSCLC with no EGFR or ALK genomic tumor aberrations in May 2020.4,7
With the approval of both Checkmate 227 and Checkmate 9LA regimens as first-line therapies, I am pleased to be able to offer metastatic NSCLC patients with additional options. Checkmate 227 provides appropriate mNSCLC patients with a chemo-free, dual I-O option with long-term, durable survival. Additionally, the Checkmate 9LA regimen with dual I-O plus limited chemo† has shown superior OS, and consistent OS, regardless of PD-L1 expression in recurrent/metastatic NSCLC patients.4,18
*In Checkmate 227, patients in the comparator arm received up to 4 cycles of platinum-doublet chemo q3w; NSQ: pemetrexed + carboplatin or cisplatin, with optional pemetrexed maintenance following chemo; SQ: gemcitabine + carboplatin or cisplatin.4,16,17
†In Checkmate 9LA, patients received 2 cycles of platinum-doublet chemo q3w in the experimental arm, and up to 4 cycles in the comparator arm; NSQ: pemetrexed + carboplatin or cisplatin (optional pemetrexed maintenance therapy in comparator arm only); SQ: paclitaxel + carboplatin.4
IMPORTANT SAFETY INFORMATION
Severe and Fatal Immune-Mediated Adverse Reactions
Immune-mediated adverse reactions listed herein may not include all possible severe and fatal immune-mediated adverse reactions.
Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue. While immune-mediated adverse reactions usually manifest during treatment, they can also occur after discontinuation of OPDIVO or YERVOY. Early identification and management are essential to ensure safe use of OPDIVO and YERVOY. Monitor for signs and symptoms that may be clinical manifestations of underlying immune-mediated adverse reactions. Evaluate clinical chemistries including liver enzymes, creatinine, adrenocorticotropic hormone (ACTH) level, and thyroid function at baseline and periodically during treatment with OPDIVO and before each dose of YERVOY. In cases of suspected immune-mediated adverse reactions, initiate appropriate workup to exclude alternative etiologies, including infection. Institute medical management promptly, including specialty consultation as appropriate.
Withhold or permanently discontinue OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information). In general, if OPDIVO or YERVOY interruption or discontinuation is required, administer systemic corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent) until improvement to Grade 1 or less. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Consider administration of other systemic immunosuppressants in patients whose immune-mediated adverse reactions are not controlled with corticosteroid therapy. Toxicity management guidelines for adverse reactions that do not necessarily require systemic steroids (e.g., endocrinopathies and dermatologic reactions) are discussed below.
Immune-Mediated Pneumonitis
OPDIVO and YERVOY can cause immune-mediated pneumonitis. The incidence of pneumonitis is higher in patients who have received prior thoracic radiation. In NSCLC patients receiving OPDIVO 3 mg/kg every 2 weeks with YERVOY 1 mg/kg every 6 weeks, immune-mediated pneumonitis occurred in 9% (50/576) of patients, including Grade 4 (0.5%), Grade 3 (3.5%), and Grade 2 (4.0%). Four patients (0.7%) died due to pneumonitis.
Immune-Mediated Colitis
OPDIVO and YERVOY can cause immune-mediated colitis, which may be fatal. A common symptom included in the definition of colitis was diarrhea. Cytomegalovirus (CMV) infection/reactivation has been reported in patients with corticosteroid-refractory immune-mediated colitis. In cases of corticosteroid-refractory colitis, consider repeating infectious workup to exclude alternative etiologies.
Immune-Mediated Hepatitis
OPDIVO and YERVOY can cause immune-mediated hepatitis.
Immune-Mediated Endocrinopathies
OPDIVO and YERVOY can cause primary or secondary adrenal insufficiency, immune-mediated hypophysitis, immune-mediated thyroid disorders, and Type 1 diabetes mellitus, which can present with diabetic ketoacidosis. Withhold OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information). For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Hypophysitis can present with acute symptoms associated with mass effect such as headache, photophobia, or visual field defects. Hypophysitis can cause hypopituitarism; initiate hormone replacement as clinically indicated. Thyroiditis can present with or without endocrinopathy. Hypothyroidism can follow hyperthyroidism; initiate hormone replacement or medical management as clinically indicated. Monitor patients for hyperglycemia or other signs and symptoms of diabetes; initiate treatment with insulin as clinically indicated.
Immune-Mediated Nephritis with Renal Dysfunction
OPDIVO and YERVOY can cause immune-mediated nephritis.
Immune-Mediated Dermatologic Adverse Reactions
OPDIVO can cause immune-mediated rash or dermatitis. Exfoliative dermatitis, including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug rash with eosinophilia and systemic symptoms (DRESS) has occurred with PD-1/PD-L1 blocking antibodies. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate nonexfoliative rashes.
YERVOY can cause immune-mediated rash or dermatitis, including bullous and exfoliative dermatitis, SJS, TEN, and DRESS. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate non-bullous/exfoliative rashes.
Withhold or permanently discontinue OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information).
Other Immune-Mediated Adverse Reactions
The following clinically significant immune-mediated adverse reactions occurred at an incidence of <1% (unless otherwise noted) in patients who received OPDIVO monotherapy or OPDIVO in combination with YERVOY or were reported with the use of other PD-1/PD-L1 blocking antibodies. Severe or fatal cases have been reported for some of these adverse reactions: cardiac/vascular: myocarditis, pericarditis, vasculitis; nervous system: meningitis, encephalitis, myelitis and demyelination, myasthenic syndrome/myasthenia gravis (including exacerbation), Guillain-Barré syndrome, nerve paresis, autoimmune neuropathy; ocular: uveitis, iritis, and other ocular inflammatory toxicities can occur; gastrointestinal: pancreatitis to include increases in serum amylase and lipase levels, gastritis, duodenitis; musculoskeletal and connective tissue: myositis/polymyositis, rhabdomyolysis, and associated sequelae including renal failure, arthritis, polymyalgia rheumatica; endocrine: hypoparathyroidism; other (hematologic/immune): hemolytic anemia, aplastic anemia, hemophagocytic lymphohistiocytosis (HLH), systemic inflammatory response syndrome, histiocytic necrotizing lymphadenitis (Kikuchi lymphadenitis), sarcoidosis, immune thrombocytopenic purpura, solid organ transplant rejection.
In addition to the immune-mediated adverse reactions listed above, across clinical trials of YERVOY monotherapy or in combination with OPDIVO, the following clinically significant immune-mediated adverse reactions, some with fatal outcome, occurred in <1% of patients unless otherwise specified: nervous system: autoimmune neuropathy (2%), myasthenic syndrome/myasthenia gravis, motor dysfunction; cardiovascular: angiopathy, temporal arteritis; ocular: blepharitis, episcleritis, orbital myositis, scleritis; gastrointestinal: pancreatitis (1.3%); other (hematologic/immune): conjunctivitis, cytopenias (2.5%), eosinophilia (2.1%), erythema multiforme, hypersensitivity vasculitis, neurosensory hypoacusis, psoriasis.
Some ocular IMAR cases can be associated with retinal detachment. Various grades of visual impairment, including blindness, can occur. If uveitis occurs in combination with other immune-mediated adverse reactions, consider a Vogt-Koyanagi-Harada–like syndrome, which has been observed in patients receiving YERVOY, as this may require treatment with systemic corticosteroids to reduce the risk of permanent vision loss.
Infusion-Related Reactions
OPDIVO and YERVOY can cause severe infusion-related reactions. Discontinue OPDIVO and YERVOY in patients with severe (Grade 3) or life-threatening (Grade 4) infusion-related reactions. Interrupt or slow the rate of infusion in patients with mild (Grade 1) or moderate (Grade 2) infusion-related reactions.
Complications of Allogeneic Hematopoietic Stem Cell Transplantation
Fatal and other serious complications can occur in patients who receive allogeneic hematopoietic stem cell transplantation (HSCT) before or after being treated with OPDIVO or YERVOY. Transplant-related complications include hyperacute graft-versus-host-disease (GVHD), acute GVHD, chronic GVHD, hepatic veno-occlusive disease (VOD) after reduced intensity conditioning, and steroid-requiring febrile syndrome (without an identified infectious cause). These complications may occur despite intervening therapy between OPDIVO or YERVOY and allogeneic HSCT.
Follow patients closely for evidence of transplant-related complications and intervene promptly. Consider the benefit versus risks of treatment with OPDIVO and YERVOY prior to or after an allogeneic HSCT.
Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, OPDIVO and YERVOY can cause fetal harm when administered to a pregnant woman. The effects of YERVOY are likely to be greater during the second and third trimesters of pregnancy. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with OPDIVO and YERVOY and for at least 5 months after the last dose.
Increased Mortality in Patients with Multiple Myeloma when OPDIVO is Added to a Thalidomide Analogue and Dexamethasone
In randomized clinical trials in patients with multiple myeloma, the addition of OPDIVO to a thalidomide analogue plus dexamethasone resulted in increased mortality. Treatment of patients with multiple myeloma with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials.
Lactation
There are no data on the presence of OPDIVO or YERVOY in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment and for 5 months after the last dose.
Serious Adverse Reactions
In Checkmate 227, serious adverse reactions occurred in 58% of patients (n=576). The most frequent (≥2%) serious adverse reactions were pneumonia, diarrhea/colitis, pneumonitis, hepatitis, pulmonary embolism, adrenal insufficiency, and hypophysitis. Fatal adverse reactions occurred in 1.7% of patients; these included events of pneumonitis (4 patients), myocarditis, acute kidney injury, shock, hyperglycemia, multi-system organ failure, and renal failure. In Checkmate 9LA, serious adverse reactions occurred in 57% of patients (n=358). The most frequent (>2%) serious adverse reactions were pneumonia, diarrhea, febrile neutropenia, anemia, acute kidney injury, musculoskeletal pain, dyspnea, pneumonitis, and respiratory failure. Fatal adverse reactions occurred in 7 (2%) patients, and included hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.
Common Adverse Reactions
In Checkmate 227, the most common (≥20%) adverse reactions were fatigue (44%), rash (34%), decreased appetite (31%), musculoskeletal pain (27%), diarrhea/colitis (26%), dyspnea (26%), cough (23%), hepatitis (21%), nausea (21%), and pruritus (21%). In Checkmate 9LA, the most common (>20%) adverse reactions were fatigue (49%), musculoskeletal pain (39%), nausea (32%), diarrhea (31%), rash (30%), decreased appetite (28%), constipation (21%), and pruritus (21%).
Please see U.S. Full Prescribing Information for OPDIVO and YERVOY:
• https://packageinserts.bms.com/pi/pi_opdivo.pdf
• https://packageinserts.bms.com/pi/pi_yervoy.pdf
References:
1. Key statistics for lung cancer. American Cancer Society. Reviewed October 1, 2019. Revised January 8, 2020. Accessed October 7, 2020. https://www.cancer.org/cancer/lung-cancer/about/key-statistics.html.
2. Lung and bronchus cancer - cancer stat facts. National Cancer Institute. Accessed October 7, 2020. https://seer.cancer.gov/statfacts/html/lungb.html.
3. Howlader N, Forjaz G, Mooradian MJ, et al. The effect of advances in lung-cancer treatment on population mortality. N Engl J Med. 2020;383:640-649.
4. OPDIVO [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
5. FDA approval for Checkmate 227. Accessed October 12, 2020. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-nivolumab-plus-ipilimumab-first-line-mnsclc-pd-l1-tumor-expression-1.
6. YERVOY [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
7. FDA approval for Checkmate 9LA. Accessed October 12, 2020. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-nivolumab-plus-ipilimumab-and-chemotherapy-first-line-treatment-metastatic-nsclc.
8. Weber JS, Hamid O, Chasalow SD, et al. Ipilimumab increases activated T cells and enhances humoral immunity in patients with advanced melanoma. J Immunother. 2012;35:89-97.
9. Farber DL, Yudanin NA, and Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14(1):24-35.
10. Ansell SM, Hurvitz SA, Koenig PA, et al. Phase I study of ipilimumab, an anti–CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non–Hodgkin lymphoma. Clin Cancer Res. 2009;15(20):6446-6453.
11. Felix J, Lambert J, Roelens M, et al. Ipilimumab reshapes T cell memory subsets in melanoma patients with clinical response. Oncoimmunology. 2016;5(7):e1136045.
12. Pedicord VA, Montalvo W, Leiner IM, and Allison JP. Single dose of anti–CTLA-4 enhances CD8+ T-cell memory formation, function, and maintenance. Proc Natl Acad Sci USA. 2011;108(1):266-271.
13. de Coaña YP, Wolodarski M, Poschke I, et al. Ipilimumab treatment decreases monocytic MDSCs and increases CD8 effector memory T cells in long-term survivors with advanced melanoma. Oncotarget. 2017;8(13):21539-21553.
14. Buchbinder EI and Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98-106.
15. Ramalingam S, Ciuleanu T-E, Pluzanski A, et al. Nivolumab + ipilimumab versus platinum-doublet chemotherapy as first-line treatment for advanced non-small cell lung cancer: Three-year update from Checkmate 227 Part 1. Oral presentation at ASCO 2020. Abstract 9500.
16. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. 2019;381:2020-2031.
17. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. 2019;381:2020-2031. [supplementary appendix].
18. Reck M, Ciuleanu T-E, Cobo M, et al. Nivolumab + ipilimumab + 2 cycles of platinum-doublet chemotherapy vs 4 cycles chemotherapy as first-line treatment for stage IV/recurrent NSCLC: Checkmate 9LA. Oral presentation at ASCO 2020. Abstract 9501.
19. Data on file. NIVO 566. Princeton, NJ: Bristol-Myers Squibb Company.
Challenges and Unmet Needs in Squamous Non-Small Cell Lung Cancer
Written by Dr. Irfan A. Mirza
This article is sponsored and developed by Boehringer Ingelheim Pharmaceuticals
Significant strides have been made in the last decade for systemic treatment options for stage IV non-small cell lung cancer (NSCLC), including those tailored for squamous and non-squamous histology.1,2 While non-squamous NSCLC has benefited from advances such as the introduction of personalized, genotyped-directed therapies, and immunotherapy drugs, the treatment options for squamous cell NSCLC remain limited.1,2
Historically, the NCCN guidelines recommended the use of platinum-based chemotherapy in the first line setting, followed by immunotherapy in the second-line.3 However, following the results of the KEYNOTE-407 study, immunotherapy together with platinum doublet chemotherapy is now recommended in the first-line setting.4,5 This leaves an unmet need for patients with metastatic squamous NSCLC who have progressed, where most treatments consist of chemotherapy.2,6
Afatinib is an oral, non-chemotherapy option for patients with metastatic squamous NSCLC who have progressed on platinum-based chemotherapy.7 Afatinib is an irreversible second-generation epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitor that selectively inhibits homo- and hetero-dimers of the ErbB receptor family (EGFR, ErbB2, and ErbB4).7
LUX-Lung 8 was a multicenter, open label, phase 3, randomized, controlled trial across 23 countries that enrolled 795 patients with advanced (stage III B and stage IV) squamous NSCLC, progressing after at least 4 cycles of platinum-based chemotherapy.8 Patients were randomized (1:1) to either afatinib 40 mg daily or erlotinib 150 mg daily until disease progression.8 The primary endpoint was progression-free survival (PFS) as assessed by an independent review committee (IRC), using RECIST v1.1 and secondary endpoints included overall survival (OS) and objective response rates as assessed by an IRC.8
In LUX-Lung 8, significant improvement in PFS and overall survival was observed for afatinib compared with erlotinib.8 The median PFS was reported as 2.4 months with afatinib and 1.9 months with erlotinib [HR, 0.82 (95% CI 0.68-0.99)] (Figure 1).8

After a median follow up of 18.4 months, median OS was 7.9 months in the afatinib group and 6.8 months in the erlotinib group [HR 0.81 (95% CI 0.69-0.95), p = 0.0077].8 Estimates of OS among patients treated with afatinib were 64% at 6 months, 36% at 1 year, and 22% at 18 months (Figure 2).8
More than half (51%) of patients treated with afatinib were able to achieve disease control (defined as complete response, partial response, stable disease, or non-complete response and non-progressive disease) compared with 40% with erlotinib.8 Excluding patients with non-complete response and non-progressive disease, disease control with afatinib was 37%, vs 29% with erlotinib, in a post hoc analysis.8 The median duration of objective response was 7.3 months with afatinib and 3.7 months with erlotinib.8
The most common adverse effects associated with afatinib were diarrhea, rash/acneiform dermatitis, stomatitis, decreased appetite, nausea, vomiting, paronychia, and pruritus.8,9 Twenty percent of patients discontinued afatinib treatment due to adverse reactions, with the most frequent adverse reactions leading to discontinuation being diarrhea in 4.1% of patients and rash/acne in 2.6%.9 Serious adverse reactions occurred in 44% of patients, with pneumonia (6.6%), diarrhea (4.6%), dehydration, and dyspnea (3.1% each) being the most frequent.9 Fatal adverse reactions in afatinib-treated patients included interstitial lung disease, pneumonia, respiratory failure, acute renal failure, and general physical health deterioration, all occurring in less than 1% of patients.9
Adverse Reactions (ARs) Reported in ≥10% of GILOTRIF-Treated Patients in LUX-Lung 89*:
GILOTRIF (n=392), erlotinib (n=395) – All Grades & Grades 3-4 ARs
Gastrointestinal Disorders
Diarrhea – GILOTRIF all grades: 75%; grades 3-4: 11%; erlotinib all grades: 41%, grades 3-4: 3%
Stomatitis† – GILOTRIF all grades: 30%; grades 3-4: 4%; erlotinib all grades: 11%, grades 3-4: 1%
Nausea – GILOTRIF all grades: 21%; grades 3-4: 2%; erlotinib all grades: 16%, grades 3-4: 1%
Vomiting – GILOTRIF all grades: 13%; grades 3-4: 1%; erlotinib all grades: 10%, grades 3-4: 1%
Skin and Subcutaneous tissue disorders
Rash/acneform dermatitis‡ – GILOTRIF all grades: 70%; grades 3-4: 7%; erlotinib all grades: 70%, grades 3-4: 11%
Pruritus – GILOTRIF all grades: 10%; grades 3-4: 0%; erlotinib all grades: 13%, grades 3-4: 0%
Metabolism and nutrition disorders
Decreased appetite – GILOTRIF all grades: 25%; grades 3-4: 3%; erlotinib all grades: 13%, grades 3-4: 0%
Infections
Paronychia§ – GILOTRIF all grades: 11%; grades 3-4: 1%; erlotinib all grades: 5%, grades 3-4: 0%
*NCI CTCAE v 3.0
† Includes stomatitis, aphthous stomatitis, mucosal inflammation, mouth ulceration, oral mucosa erosion, mucosal erosion, mucosal ulceration
‡ Includes acne, dermatitis, acneiform dermatitis, eczema, erythema, exfoliative rash, folliculitis, rash, rash generalized, rash macular, rash maculo-papular,
rash pruritic, rash pustular, skin exfoliation, skin fissures, skin lesion, skin reaction, skin toxicity, skin ulcer
§ Includes paronychia, nail infection, nail bed infection
In summary, LUX-Lung 8 met its primary and secondary endpoints and remains the largest prospective head-to-head trial that compares two TKIs for second-line treatment of patients with squamous NSCLC.8 Future studies should focus on understanding the clinical profile of afatinib within the context of other commonly-used treatment modalities, such as chemotherapy. In a disease setting with few treatment options, and a pandemic which can make delivery of infusions challenging, afatinib offers patients with metastatic squamous NSCLC an opportunity to receive a chemotherapy-free, oral option once they have progressed following treatment with standard, platinum based, first line treatment.8,9
INDICATIONS AND USAGE
GILOTRIF is indicated for the treatment of patients with metastatic squamous NSCLC progressing after platinum-based chemotherapy.
IMPORTANT SAFETY INFORMATION FOR GILOTRIF® (afatinib) TABLETS
WARNINGS AND PRECAUTIONS
Diarrhea
• GILOTRIF can cause diarrhea which may be severe and can result in dehydration with or without renal impairment. In clinical studies, some of these cases were fatal.
• For patients who develop Grade 2 diarrhea lasting more than 48 hours or Grade 3 or greater diarrhea, withhold GILOTRIF until diarrhea resolves to Grade 1 or less, and then resume at a reduced dose.
• Provide patients with an anti-diarrheal agent (e.g., loperamide) for self-administration at the onset of diarrhea and instruct patients to continue anti-diarrheal until loose stools cease for 12 hours.
Bullous and Exfoliative Skin Disorders
• GILOTRIF can result in cutaneous reactions consisting of rash, erythema, and acneiform rash. In addition, palmar-plantar erythrodysesthesia syndrome was observed in clinical trials in patients taking GILOTRIF.
• Discontinue GILOTRIF in patients who develop life-threatening bullous, blistering, or exfoliating skin lesions. For patients who develop Grade 2 cutaneous adverse reactions lasting more than 7 days, intolerable Grade 2, or Grade 3 cutaneous reactions, withhold GILOTRIF. When the adverse reaction resolves to Grade 1 or less, resume GILOTRIF with appropriate dose reduction.
• Postmarketing cases of toxic epidermal necrolysis (TEN) and Stevens Johnson syndrome (SJS) have been reported in patients receiving GILOTRIF. Discontinue GILOTRIF if TEN or SJS is suspected.
Interstitial Lung Disease
• Interstitial Lung Disease (ILD) or ILD-like adverse reactions (e.g., lung infiltration, pneumonitis, acute respiratory distress syndrome, or alveolitis allergic) occurred in patients receiving GILOTRIF in clinical trials. In some cases, ILD was fatal. The incidence of ILD appeared to be higher in Asian patients as compared to white patients.
• Withhold GILOTRIF during evaluation of patients with suspected ILD, and discontinue GILOTRIF in patients with confirmed ILD.
Hepatic Toxicity
• Hepatic toxicity as evidenced by liver function tests abnormalities has been observed in patients taking GILOTRIF. In 4257 patients who received GILOTRIF across clinical trials, 9.7% had liver test abnormalities, of which 0.2% were fatal.
• Obtain periodic liver testing in patients during treatment with GILOTRIF. Withhold GILOTRIF in patients who develop worsening of liver function. Discontinue treatment in patients who develop severe hepatic impairment while taking GILOTRIF.
Gastrointestinal Perforation
• Gastrointestinal (GI) perforation, including fatal cases, has occurred with GILOTRIF. GI perforation has been reported in 0.2% of patients treated with GILOTRIF among 3213 patients across 17 randomized controlled clinical trials.
• Patients receiving concomitant corticosteroids, nonsteroidal anti-inflammatory drugs (NSAIDs), or anti-angiogenic agents, or patients with increasing age or who have an underlying history of GI ulceration, underlying diverticular disease, or bowel metastases may be at an increased risk of perforation.
• Permanently discontinue GILOTRIF in patients who develop GI perforation.
Keratitis
• Keratitis has been reported in patients taking GILOTRIF.
• Withhold GILOTRIF during evaluation of patients with suspected keratitis. If diagnosis of ulcerative keratitis is confirmed, interrupt or discontinue GILOTRIF. If keratitis is diagnosed, the benefits and risks of continuing treatment should be carefully considered. GILOTRIF should be used with caution in patients with a history of keratitis, ulcerative keratitis, or severe dry eye. Contact lens use is also a risk factor for keratitis and ulceration.
Embryo-Fetal Toxicity
• GILOTRIF can cause fetal harm when administered to a pregnant woman. Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
• Advise females of reproductive potential to use effective contraception during treatment, and for at least 2 weeks after the last dose of GILOTRIF. Advise female patients to contact their healthcare provider with a known or suspected pregnancy.
ADVERSE REACTIONS
Adverse Reactions observed in clinical trials were as follows:
Previously Treated, Metastatic Squamous NSCLC
• In GILOTRIF-treated patients (n=392) the most common adverse reactions (≥20% all grades & vs erlotinib-treated patients (n=395)) were diarrhea (75% vs 41%), rash/acneiform dermatitis (70% vs 70%), stomatitis (30% vs 11%), decreased appetite (25% vs 26%), and nausea (21% vs 16%).
• Serious adverse reactions were reported in 44% of patients treated with GILOTRIF. The most frequent serious adverse reactions reported in patients treated with GILOTRIF were pneumonia (6.6%), diarrhea (4.6%), and dehydration and dyspnea (3.1% each). Fatal adverse reactions in GILOTRIF-treated patients included ILD (0.5%), pneumonia (0.3%), respiratory failure (0.3%), acute renal failure (0.3%), and general physical health deterioration (0.3%).
DRUG INTERACTIONS
Effect of P-glycoprotein (P-gp) Inhibitors and Inducers
• Concomitant use of P-gp inhibitors (including but not limited to ritonavir, cyclosporine A, ketoconazole, itraconazole, erythromycin, verapamil, quinidine, tacrolimus, nelfinavir, saquinavir, and amiodarone) with GILOTRIF can increase exposure to afatinib.
• Concomitant use of P-gp inducers (including but not limited to rifampicin, carbamazepine, phenytoin, phenobarbital, and St. John’s wort) with GILOTRIF can decrease exposure to afatinib.
USE IN SPECIFIC POPULATIONS
Lactation
• Because of the potential for serious adverse reactions in breastfed infants from GILOTRIF, advise women not to breastfeed during treatment with GILOTRIF and for 2 weeks after the final dose.
Females and Males of Reproductive Potential
• GILOTRIF may reduce fertility in females and males of reproductive potential. It is not known if the effects on fertility are reversible.
Renal Impairment
• Patients with severe renal impairment (estimated glomerular filtration rate [eGFR] 15 to 29 mL/min/1.73 m2) have a higher exposure to afatinib than patients with normal renal function. Administer GILOTRIF at a starting dose of 30 mg once daily in patients with severe renal impairment. GILOTRIF has not been studied in patients with eGFR <15 mL/min/1.73 m2 or who are on dialysis.
Hepatic Impairment
• GILOTRIF has not been studied in patients with severe (Child Pugh C) hepatic impairment. Closely monitor patients with severe hepatic impairment and adjust GILOTRIF dose if not tolerated.
REFERENCES
1. Baxevanos P, Mountzios G. Novel chemotherapy regimens for advanced lung cancer: have we reached a plateau? Ann Transl Med. 2018;6(8):139.
2. Santos ES, Hart L. Advanced Squamous Cell Carcinoma of the Lung: Current Treatment Approaches and the Role of Afatinib. Onco Targets Ther. 2020 Sep 22;13:9305-9321.
3. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer. V.1.2016. ©National Comprehensive Cancer Network, Inc. 2016. All rights reserved. Accessed November 2, 2020. To view the most recent and complete version of the guidelines, go online to NCCN.org.
4. Paz-Ares L, et al. Pembrolizumab plus Chemotherapy for Squamous NSCLC. N Engl J Med. 2018;379: 2040-2051; DOI:10.1056/NEJMoa1810865
5. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer. V.8.2020. ©National Comprehensive Cancer Network, Inc. 2020. All rights reserved. Accessed November 2, 2020. To view the most recent and complete version of the guidelines, go online to NCCN.org.
6. Paik PK, et al. New treatment options in advanced squamous cell lung cancer. Am Soc Clin Oncol Educ Book. 2019;39:e198-e206.
7. Hirsh V. Next-Generation Covalent Irreversible Kinase Inhibitors in NSCLC: Focus on Afatinib. BioDrugs. 2015;29(3):167 183.
8. Soria JC, et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2015;16(8):897 907.
9. GILOTRIF [prescribing information]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc.
Please review the Full Prescribing Information and Patient Information at www.gilotrifhcp.com
Adjuvant TAGRISSO® in Resected EGFR-Mutated Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R substitution mutation in Exon 21. Approximately 25% of patients with EGFR mutated NSCLC have brain metastases at diagnosis, increasing to approximately 40% within two years of diagnosis. The presence of brain metastases often reduces median survival to less than eight months. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to the most common, acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients.
TAGRISSO® (Osimertinib) is a highly selective third-generation Epidermal Growth Factor Receptor (EGFR) TKI presently approved by the FDA, for the first-line treatment of patients with metastatic NSCLC, whose tumors have Exon 19 deletions or Exon 21 L858R mutations, as well as treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, whose disease has progressed on or after EGFR-TKI therapy. Further, TAGRISSO® has higher CNS penetration and is therefore able to induce responses in 70-90% of patients with brain metastases. Among patients with metastatic, EGFR-mutant NSCLC, first-line treatment with TAGRISSO® significantly improved median Overall Survival, compared with TARCEVA® and IRESSA®, and should therefore be considered the preferred regimen.
Surgical resection is the primary treatment for approximately 30% of patients with NSCLC who present with early Stage (I–IIIA) disease. These patients are often treated with platinum-based adjuvant chemotherapy to decrease the risk of recurrence. Nonetheless, 45-75% of these patients develop recurrent disease. There is therefore an unmet need for this patient population.
ADAURA is a global, double-blind, randomized Phase III study, which assessed the efficacy and safety of TAGRISSO® versus placebo in patients with Stage IB–IIIA EGFR mutated NSCLC, after complete tumor resection and adjuvant chemotherapy, when indicated. In this study, 682 patients with completely resected Stage IB, II, IIIA NSCLC, with or without postoperative adjuvant chemotherapy, were randomly assigned 1:1 to receive either TAGRISSO® 80 mg orally once daily (N=339) or placebo (N=343) once daily, for up to 3 years. Eligible patients had an ECOG Performance Status of 0 or 1, with confirmed EGFR mutations (Exon 19del or L858R). Treatment groups were well balanced and patients were stratified by Stage (IB/II/IIIA), mutation type (Exon 19del/L858R), and race (Asian/non-Asian).
Most patients with Stage II to IIIA disease (76%) and approximately a quarter of the patients with Stage IB disease (26%) received adjuvant platinum-based chemotherapy. The Primary endpoint was Disease Free Survival (DFS) in Stage II–IIIA patients. Secondary endpoints included DFS in the overall population of patients with Stage IB to IIIA disease, Overall Survival (OS) and safety. Following Independent Data Monitoring Committee recommendation, the trial was unblinded early, due to efficacy. The authors reported the results from the unplanned interim analysis.
It was noted that in the patients with Stage II/IIIA disease, the DFS had not been reached with TAGRISSO® versus 19.6 months with placebo (HR=0.17; P<0.001). This was equal to an 83% reduction in the risk of recurrence or death, indicating a significantly longer DFS among patients in the TAGRISSO® group, compared to those in the placebo group. The 2-year DFS rate in this patient group with TAGRISSO® was 90% versus 44% with placebo.
In the overall population, which included Stage IB to IIIA disease, the median DFS was not reached with TAGRISSO® versus 27.5 months with placebo (HR=0.20; P<0.001). This Hazard Ratio equaled an 80% reduction in the risk of disease recurrence or death among patients in the TAGRISSO® group compared to those in the placebo group. The 2-year DFS rate in the overall population was 89% with TAGRISSO® versus 52% with placebo.
The benefit favoring TAGRISSO® with respect to DFS was observed consistently across all predefined subgroups including disease Stages IB, II, and IIIA and use or nonuse of adjuvant chemotherapy. The benefit with TAGRISSO® was greater at more advanced stages of disease (among patients with Stage IIIA disease, the overall HR was 0.12, among those with Stage II disease, the HR was 0.17, and among those with Stage IB disease, the HR was 0.39). At 2 years, 98% of the patients in the TAGRISSO® group and 85% of the patients in the placebo group were alive without CNS-related disease (HR for CNS disease recurrence or death=0.18). This indicated an 82% reduction in the risk of CNS disease recurrence or death with TAGRISSO®. The Overall Survival data were immature at the time of this interim analysis. Adverse Events were consistent with the known safety profile of TAGRISSO®.
The authors concluded that adjuvant TAGRISSO® is the first targeted agent in a global randomized trial, to show a statistically significant and clinically meaningful improvement in Disease Free Survival, among patients with Stage IB/II/IIIA EGFR mutation-positive NSCLC, and provides an effective new treatment strategy for this patient group.
Osimertinib in Resected EGFR-Mutated Non-Small-Cell Lung Cancer. Wu Y-L, Tsuboi M, He J, et al. for the ADAURA Investigators. N Engl J Med 2020; 383:1711-1723.
GILOTRIF® in EGFR Positive Non Small Cell Lung Cancer Harboring Uncommon Mutations
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients, and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations. The majority of patients have classical EGFR mutations which are either Exon 19 deletions or L858R substitution mutation in Exon 21, and for those patients with EGFR mutation-positive NSCLC, EGFR-TKIs are the first choice of treatment. However, around 5-20% of tumors harbor Major uncommon mutations, such as G719X, L861Q and S768I, as well as other more rare mutations, and these uncommon EGFR mutations show heterogeneity in their response to EGFR-TKIs. Compared with other EGFR mutations, G719X, L861Q and S768I substitution mutations are associated with a poorer prognosis and have limited treatment options.
GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. GILOTRIF® demonstrated clinical activity against Major uncommon EGFR mutations such as G719X, L861Q and S768I which is more often seen in Asian patients, and is FDA approved in this setting. There are however, few clinical data regarding the efficacy of the available EGFR-TKIs against other uncommon EGFR mutations, and there is no knowledge of ethnic differences in prevalence and outcomes.
This study investigated the efficacy of GILOTRIF® in EGFR mutation positive NSCLC among Asian and non-Asian patients with uncommon mutations. Uncommon mutations were classed into five categories- Major uncommon (G719X, L861Q and S768I), Compound, Exon 20 insertions, T790M Mutation, and Other. Patients may have more than uncommon mutation.
The researchers conducted a pooled analysis from randomized clinical trials and Real-World Studies and examined the activity of GILOTRIF® in Asian and non-Asian patients with NSCLC and uncommon EGFR mutations, who had not received prior treatment with EGFR TKIs. All identified patients included in this study had outcome data such as Time to Treatment failure (TTF) or Objective Response Rate (ORR) available. The total number of evaluable patients were 298 (N=298), of whom 60% were Asian (N=178) and 40% were Non-Asian (N=120). The median patient age ranged from 60-66 years across the different mutation groups. Approximately 40% of patients had Major uncommon mutations such as G719X, L861Q and S768I, 24% had Exon 20 insertions, 12% had T790M mutations and 24% had Compound and Other mutations. When broken by ethnicity, among Asian patients, approximately 62% had Major uncommon mutations, 14% had Compound mutations and 16% had Exon 20 insertions. Among non-Asian patients, 35% had Major uncommon mutations, approximately 7% had Compound mutations and 39% had Exon 20 insertions. The Endpoints included Objective Response Rate (ORR), Duration of Response (DoR) and Time to Treatment Failure (TTF), and outcomes were compared in Asian and non-Asian EGFR-TKI-naïve patients.
This analysis showed that the efficacy of GILOTRIF® was unaffected by ethnicity, and the Overall Response Rate (ORR) among tumors with Major uncommon mutations was 66% in Asian patients versus 59% in non-Asian patients, and the median Duration of Response (DoR) was 14.7 months compared with 15.9 months respectively. Among those with Major uncommon mutations, the ORR in tumors harboring G719X mutation was 62% in Asian patients and 65% in non-Asian patients. Among those tumors with a L861Q mutation, the ORRs were 60% versus 50%, respectively and among those with a S768I mutation, the ORRs were 80% versus 25%, respectively. The Overall Response Rate (ORR) among tumors with Compound mutations was 81% in Asian patients versus 100% in non-Asian patients and the median Duration of Response (DoR) was 11.5 months compared with 18.6 months respectively. Among patients who harbored Exon 20 insertions, the ORR with GILOTRIF® in Asian patients was 21% versus 23% in non-Asian patients, with a Duration of Response of 11 months and 10.7 months, respectively.
It was concluded that GILOTRIF® shows clinical activity against uncommon EGFR mutations in both Asian and non-Asian patients, with durable clinical responses, and should be considered as a first-line treatment option in Asian and non-Asian patients with Major uncommon (G719X, L861Q and S768I) and Compound EGFR mutations.
Afatinib in Asian and non-Asian patients (pts) with EGFR mutation-positive (EGFRm+) NSCLC harboring major uncommon mutations. Yang JC-H, Schuler M, Popat S, et al. Presented at: 2020 IASLC North America Conference on Lung Cancer; October 16-17, 2020; Virtual. Abstract MO01.36.
KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors
SUMMARY: The KRAS (Kirsten rat sarcoma viral oncogene homologue) proto-oncogene encodes a protein that is a member of the small GTPase super family. The KRAS gene provides instructions for making the KRAS protein, which is a part of a signaling pathway known as the RAS/MAPK pathway. By relaying signals from outside the cell to the cell nucleus, the protein instructs the cell to grow, divide and differentiate. The KRAS protein is a GTPase, and converts GTP into GDP. To transmit signals, the KRAS protein must be turned on, by binding to a molecule of GTP. When GTP is converted to GDP, the KRAS protein is turned off or inactivated, and when the KRAS protein is bound to GDP, it does not relay signals to the cell nucleus. The KRAS gene is in the Ras family of oncogenes, which also includes two other genes, HRAS and NRAS. When mutated, oncogenes have the potential to change normal cells cancerous.
KRAS is the most frequently mutated oncogene in human cancers and are often associated with resistance to targeted therapies and poor outcomes. The KRAS-G12C mutation occurs in approximately 12-15% of Non Small Cell Lung Cancers (NSCLC) and in 3-5% of Colorectal cancers and other solid cancers. G12C is a single point mutation with a Glycine-to-Cysteine substitution at codon 12. This substitution favors the activated state of KRAS, resulting in a predominantly GTP-bound KRAS oncoprotein, amplifying signaling pathways that lead to oncogenesis.
Sotorasib (AMG 510) is a small molecule that specifically and irreversibly inhibits KRAS-G12C and traps KRAS-G12C in the inactive GDP-bound state. Preclinical studies in animal models showed that Sotorasib inhibited nearly all detectable phosphorylation of Extracellular signal-Regulated Kinase (ERK), a key downstream effector of KRAS, leading to durable complete regression of KRAS-G12C tumors.
The authors conducted a multicenter, open label Phase I trial of Sotorasib, in patients with advanced solid tumors harboring the KRAS-G12C mutation. This trial consisted of dose escalation and expansion cohorts and included a total of 129 patients, of whom 59 patients had NSCLC, 42 had Colorectal cancer, and 28 patients had other tumor types (Appendiceal, Endometrial, Pancreatic cancers and Melanoma). Sotorasib was administered orally once daily and each treatment cycle was 21 days. The planned dose levels for the escalation cohorts were 180, 360, 720, and 960 mg. Treatment was continued until disease progression or unacceptable toxicity. The median patient age was 62 years and most of the enrolled patients were heavily pretreated and had received a median of 3 previous lines of anticancer therapy for metastatic disease. Among the NSCLC patient cohort, approximately 90% of patients were current or former smokers and had received anti- Programmed cell Death protein-1 (PD-1) or PD-Ligand 1 (PD-L1) therapies. All patients had received previous platinum-based chemotherapy. The Primary endpoint was safety, including the incidence of dose-limiting toxicities and key Secondary end points were pharmacokinetics and Objective Response Rates. The Sotorasib dose of 960 mg daily was identified as the dose for the expansion cohort. The median follow up was 11.7 months and the median duration of treatment was 3.9 months, with 57% of patients having received treatment for 3 months or more, and 29% of patients, for 6 months or more.
Among those patients with NSCLC, 32.2% of the patients had a confirmed Objective Response (Complete or Partial Response at all dose levels, and 88% had disease control (Objective Response or Stable disease), with a median Progression Free Survival of 6.3 months. Responses were rapid and were seen at week 6, and these responses were durable and ongoing at a median follow up of nearly a year.
Among the colorectal cancer subgroup, at a median follow up of 12.8 months, 7% had a confirmed response, and 74% had disease control, with a median duration of stable disease of 5.4 months and median PFS of 4 months. Responses were also observed in patients with Pancreatic, Endometrial, and Appendiceal cancers and Melanoma. It has been postulated that the inconsistent tumor responses noted between NSCLC and Colorectal cancer suggests either that KRAS-G12C is not the dominant oncogenic driver for colorectal cancer or that other pathways such as Wnt or EGFR pathways may mediate oncogenic signaling beyond KRAS. The authors suggest that a viable option would be to combine Sotorasib with therapies that block additional pathways, as was shown by studies in BRAF V600E-mutant Colorectal cancer. Approximately 57% of patients had treatment-related Adverse Events, of whom, about 12% had Grade 3 or 4 events. These toxicities included abnormal liver function studies, anemia, lymphopenia and diarrhea.
It was concluded Sotorasib showed promising anticancer activity in patients with heavily pretreated advanced solid tumors harboring the KRAS-G12C mutation. Studies evaluating Sotorasib as monotherapy or in combination with various agents in patients with NSCLC or other solid tumors are under way
KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. Hong DS, Fakih MG, Strickler JH, et al. N Engl J Med 2020; 383:1207-1217.
Optimal Duration of Immune Checkpoint Inhibitors in Advanced Non Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers.
Immunotherapy with Immune Checkpoint Inhibitors (ICIs) has revolutionized cancer care and has become one of the most effective treatment options, by improving Overall Response Rate and prolongation of survival, across multiple tumor types. These agents target Programmed cell Death protein 1 (PD 1), Programmed cell Death Ligand 1 (PD L1), Cytotoxic T-Lymphocyte-Associated protein 4 (CTLA 4), and many other important regulators of the immune system. Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response. Biomarkers predicting responses to ICI’s include Tumor Mutational Burden (TMB), Mismatch Repair (MMR) status, and Programmed cell Death Ligand 1 (PD L1) expression. Other biomarkers such as Tumor Infiltrating Lymphocytes (TILs), TIL derived Interferon gamma, Neutrophil to Lymphocyte ratio, and peripheral cytokines, have also been proposed as predictors of response. The optimal duration of treatment with checkpoint inhibitors across tumor types is currently unknown and finding the balance between efficacy, toxicity and cost of therapy remains an ongoing challenge. There are presently no adequately powered, prospective, checkpoint inhibitor trials, comparing different treatment durations.
OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response, and unleashing the T cells. The authors in this study explored the impact of duration of treatment with OPDIVO®, on outcomes, in patients with previously treated advanced NSCLC, in a randomized study.
CheckMate 153 is a largely community based, ongoing, Phase IIIb/IV study, reflecting a real-world population, designed to evaluate the efficacy and safety of OPDIVO® monotherapy treatment duration, in previously treated advanced NSCLC. In this study, patients with previously treated advanced or metastatic NSCLC received OPDIVO® 3 mg/kg IV every 2 weeks until disease progression, unacceptable toxicity, or for 1 year. Treatment beyond initial progressive disease was permitted for patients with investigator-assessed clinical benefit, no rapidly progressive disease, and stable ECOG performance status, who were tolerating treatment. Patients who continued to receive treatment at 1 year were randomly assigned, regardless of response status to continue OPDIVO®, or to stop treatment (1-year fixed duration group), with the option of receiving OPDIVO® retreatment on study after disease progression. The Primary end point of safety was previously reported. Exploratory post-random assignment end points were added. Safety and tolerability, Progression Free survival (PFS), Overall Survival (OS), and Objective Response Rate (ORR) were assessed from the time of random assignment of those patients who continued to receive treatment at 1 year. The comparison was between a fixed 1-year treatment regimen and continuous therapy.
Of the 1,428 patients who received OPDIVO® in this study, 252 patients were randomly assigned to continuous treatment (N=127) or 1-year fixed-duration treatment (N=125). With minimum post-random assignment follow up of 13.5 months, median PFS was longer with continuous treatment versus 1-year fixed duration treatment (24.7 months versus 9.4 months; HR=0.56). Median Overall Survival from random assignment was also longer with continuous treatment versus 1-year fixed duration treatment in the Progression-Free Survival population (Not Reached versus 32.5 months; HR, 0.61), as well as in the Intent To Treat population (Not reached versus 28.8 months; HR, 0.62). New onset treatment-related Adverse Events occurred in a few patients and no new safety signals were identified.
The authors concluded that the above findings from an exploratory analysis represent the first randomized data on continuous versus fixed-duration immunotherapy, in previously treated patients with advanced NSCLC, and suggest that continuing OPDIVO® beyond 1 year improves outcomes.
Continuous Versus 1-Year Fixed-Duration Nivolumab in Previously Treated Advanced Non–Small-Cell Lung Cancer: CheckMate 153. Waterhouse DM, Garon EB, Chandler J, et al. DOI: 10.1200/JCO.20.00131 Journal of Clinical Oncology. Published online September 10, 2020.
FDA Approves GAVRETO® for Metastatic RET Fusion-Positive Non Small Cell Lung Cancer
SUMMARY: The FDA on September 4, 2020, granted accelerated approval to GAVRETO® (Pralsetinib) for adult patients with metastatic RET fusion-positive Non Small Cell Lung Cancer (NSCLC), as detected by an FDA approved test. The FDA also approved the Oncomine Dx Target (ODxT) Test as a companion diagnostic for GAVRETO®. Lung cancer is the second most common cancer in both men and women  and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers.
and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers.
In addition to the well characterized gene fusions involving ALK and ROS1 in NSCLC, genetic alterations involving other kinases including EGFR, BRAF, RET, NTRK, are all additional established targetable drivers. These genetic alterations are generally mutually exclusive, with no more than one predominant driver in any given cancer. The hallmark of all of these genetic alterations is oncogene addiction, in which cancers are driven primarily, or even exclusively, by aberrant oncogene signaling, and are highly susceptible to small molecule inhibitors.
RET kinase is a transmembrane Receptor Tyrosine Kinase and plays an important role during the development and maintenance of a variety of tissues, including neural and genitourinary tissues. RET signaling activates downstream pathways such as JAK/STAT3 and RAS/RAF/MEK/ERK and leads to cellular proliferation, survival, invasion, and metastasis. Oncogenic alterations to the RET proto-oncogene results in uncontrolled cell growth and enhanced tumor invasiveness. RET alterations include RET rearrangements, leading to RET fusions, and activating point mutations occurring across multiple tumor types. RET fusions have been identified in approximately 2% of NSCLCs, 10-20% of non-medullary thyroid cancers. Activating RET point mutations account for approximately 60% of sporadic Medullary Thyroid Cancers (MTC) and more than 90% of inherited MTCs. Other cancers with documented RET alterations include colorectal, breast, and several hematologic malignancies.
GAVRETO® is an oral, highly potent, selective RET kinase inhibitor targeting oncogenic RET alterations, including fusions and mutations, regardless of the tissue of origin. The efficacy of GAVRETO® was investigated in a multicenter, open-label, multi-cohort, Phase I/II basket clinical trial (ARROW), in patients with tumors showing RET alterations. Identification of RET gene alterations was prospectively determined in local laboratories using either Next Generation Sequencing (NGS), Fluorescence In Situ Hybridization (FISH), or other tests. (In a basket trial, tumors with different histologies and single biomarker are placed in different baskets and receive a single treatment). The main efficacy outcome measures were Overall Response Rate (ORR) and response duration, as determined by a blinded Independent Review Committee, using RECIST criteria.
The efficacy for RET fusion-positive NSCLC was evaluated in 87 patients previously treated with platinum-based chemotherapy. Patients received GAVRETO® 400 mg orally once daily. The ORR was 57%, with a Complete Response (CR) rate of 5.7% and 80% of responding patients had responses lasting 6 months or longer. The median Duration of Response was not reached. Efficacy was also evaluated in 27 patients who never received systemic treatment and the ORR in this patient group was 70% with 11% CR rate and 58% of responding patients had responses lasting 6 months or longer. The most common adverse reactions (25% or more) were fatigue, constipation, musculoskeletal pain and hypertension.
It was concluded that patients treated with GAVRETO® had a rapid, potent, and durable clinical response, in patients with advanced RET fusion positive NSCLC, regardless of RET fusion partner, presence of brain metastases, or prior therapies.
Gainor JF, Curigliano G, Kim D-W, et al. DOI: 10.1200/JCO.2020.38.15_suppl.9515 Journal of Clinical Oncology 38, no. 15_suppl (May 20, 2020) 9515-9515.
Next-Generation Sequencing Superior to Single Gene Testing in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
In addition to the well characterized gene fusions involving ALK and ROS1 in NSCLC, genetic alterations involving other kinases including EGFR, BRAF, RET, NTRK, MET, HER2 are all additional established targetable drivers. These genetic alterations are generally mutually exclusive, with no more than one predominant driver in any given cancer. The hallmark of all of these genetic alterations is oncogene addiction, in which cancers are driven primarily, or even exclusively, by aberrant oncogene signaling, and are highly susceptible to small molecule inhibitors. Patients with nonsquamous NSCLC should therefore be tested for Actionable Driver Oncogenes, as highly effective treatments may be available for these patients. Nonetheless, Single Gene Testing for EGFR and ALK is more common in the US rather than broad multigene panel testing with Next-Generation Sequencing.
Next-Generation Sequencing (NGS) platforms or second-generation sequencing, unlike the first-generation sequencing, known as Sanger sequencing, perform massively parallel sequencing, which allows sequencing of millions of fragments of DNA from a single sample. With this high-throughput sequencing, the entire genome can be sequenced in less than 24 hours. There are a number of different NGS platforms using different sequencing technologies and NGS can be used to sequence and systematically study the cancer genomes in their entirety or specific areas of interest in the genome or small numbers of individual genes. Recently reported genomic profiling studies, performed in patients with advanced cancer suggest that actionable mutations are found in 20-40% of patients’ tumors.
The authors in this study used a decision analytic model they had developed, and compared the value of broad NGS-based testing, to Single Gene Testing (SGT), in patients with nonsquamous NSCLC, and discussed their implications for the US population. The authors noted that Single Gene Testing for EGFR and ALK is relatively common (>80%) in the US, whereas testing for less common Actionable Driver Oncogenes is rare. The broader NGS Actionable Driver Oncogene panel includes EGFR, ALK, ROS1, BRAF, RET, MET, NTRK. The authors took into consideration reimbursement by CMS for broad NGS-based testing ($627.50), reimbursement for Single Gene testing (EGFR+ALK $732.30), and the cost of treatment for 2 years at $10K/year ($20,000). The expected prevalence of Actionable Driver Oncogenes among non squamous NSCLC patients, as well as survival outcomes of patients, in the presence versus absence of an Actionable Driver Oncogenes treatment strategy, was calculated based on current literature. The number of eligible patients with nonsquamous NSCLC, for testing in the US, were 89,000 (N=89,000). The estimated number of patients with Actionable Driver Oncogenes (EGFR, ALK, ROS1, BRAF, RET, MET, NTRK) was 26,300 (N=26,300). The goal of this study was to measure the cost and value differences when one chose to run a Single Gene Testing (narrow genomics panel), which included interrogation for either EGFR or ALK, versus a broader NGS panel. The potential value of each testing approach was measured based on Life Years Gained (LYG) and the cost per LYG. (Life Years gained is a modified mortality measure where remaining life expectancy is taken into account).
It was noted that a broad NGS approach to test for genetic alterations resulted in additional Life Year Gains with cost savings, compared to Single Gene Testing for EGFR or ALK. This analytical model suggested that at the current 80% testing rate, replacing Single Gene Testing with NGS would result in an additional 21,019 Life Year Gained, with reduced cost per LYG of $599. Increasing testing from 80% to 100% of eligible patients would further increase the Life Year Gained by 15,017. If 100% of eligible patients were tested with NGS and every identified patient received treatment, the cost per Life Year Gained with this strategy would be $16,641.57.
According to this decision model, the estimated median survival and 5-year survival for a patient who was tested with NGS, followed by a highly effective therapy selected on the basis of that alteration, would be 39 months and 25%, respectively. For a patient who had an Actionable Driver Oncogene that was not identified by Single Gene Testing, the estimated median survival would be 14 months and 5-year survival would be 5%. This analysis suggested that not running broad multigene NGS panel routinely for eligible patients, and only using Single Gene Testing could be a missed opportunity, as actionable mutations would be missed and patients may not get the most effective therapy for their disease.
The authors concluded that based on their decision analytic model, when highly effective therapy is available to all identified patients with Actionable Driver Oncogenes, broad NGS testing, compared to Single Gene Testing for EGFR or ALK, leads to large gains in Life Years, at reduced cost per Life Year Gained, compared to Single Gene Testing. This model supports universal NGS testing of all patients with advanced nonsquamous NSCLC.
A model comparing the value of broad next-gen sequencing (NGS)-based testing to single gene testing (SGT) in patients with nonsquamous non-small cell lung cancer (NSCLC) in the United States. Pennell NA, Zhou J, Hobbs B. J Clin Oncol 38: 2020 (suppl; abstr 9529)
First Line CYRAMZA® Plus ERLOTINIB® for EGFR-Mutated Non Small Cell Lung Cancer
SUMMARY: The FDA on May 29, 2020 approved CYRAMZA® (Ramucirumab) in combination with TARCEVA® (Erlotinib) for first line treatment of metastatic Non Small Cell Lung Cancer (NSCLC) with Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) mutations.
Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R substitution mutation in Exon 21. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to the most common, acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients.
TAGRISSO® (Osimertinib) is a highly selective third-generation Epidermal Growth Factor Receptor (EGFR) TKI presently approved by the FDA, for the first-line treatment of patients with metastatic NSCLC, whose tumors have Exon 19 deletions or Exon 21 L858R mutations, as well as treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, whose disease has progressed on or after EGFR-TKI therapy. Previously published data from the Phase III FLAURA study showed that first-line treatment with TAGRISSO® was superior to first-line treatment with other first and second generation TKI’s, in patients with EGFR-mutated NSCLC, with improved median Overall Survival. Patients with the exon 21 L858R substitution mutation however are less sensitive to TKIs and their PFS therefore has been lower, than those with exon 19 deletions. In the FLAURA study the PFS in the exon 21 mutation group was 14.4 months compared with 21.4 months for patients with an exon 19 deletion, when treated with first line TAGRISSO®. Furthermore, widespread use of TAGRISSO® has led to acquired resistance. Evolving data has shown limited responses to immune checkpoint inhibitors after disease progression on TKIs. There is therefore a critical need to develop new TKI-based therapies, and novel treatment approaches, combining TKI’s with other targeted therapies. RELAY trial was designed to address this unmet need.
CYRAMZA® is a recombinant human monoclonal IgG1 antibody that binds to the human Vascular Endothelial Growth Factor Receptor- 2 (VEGFR-2), preventing the interaction of VEGFR-2 with its ligands. CYRAMZA® was previously approved for use in combination with TAXOTERE® (Docetaxel) for the treatment of patients with metastatic NSCLC who progressed while on or following treatment with platinum-based chemotherapy. Several preclinical studies have shown that dual blockade of the EGFR and VEGF pathways in EGFR-mutated metastatic NSCLC is synergistic, with higher antitumor activity, when compared with inhibition of the EGFR pathway alone. This has been attributed to upregulation of VEGF in the tumor microenvironment, when tumor cells harbor EGFR mutations.
RELAY trial is global, multicenter, randomized, double-blind, placebo-controlled, Phase III study in which 449 patients with previously untreated metastatic NSCLC, who harbored either an EGFR exon 19 deletion or exon 21 L858R substitution mutation, were enrolled. Patients were randomized 1:1 to receive TARCEVA® 150 mg orally daily in combination with either CYRAMZA® 10 mg/kg IV (N=224) or placebo (N=225), every 2 weeks. Treatment was continued until disease progression or unacceptable toxicity. Both treatment groups were well balanced. Patients had an ECOG performance status of 0-1, median age was 64 years, and about 60% of patients were never smokers. Those with a known EGFR T790M mutation, received prior treatment with an EGFR TKI or chemotherapy, or had brain metastases, were ineligible for study enrollment. Patients were stratified by sex, EGFR mutation type, and EGFR testing methodology. The Primary end point was Progression Free Survival (PFS). Secondary end points included Overall Survival (OS), Overall Response Rate (ORR) and Duration of Response (DOR).
At a median follow up of 20.7 months, the median PFS was 19.4 months in the CYRAMZA® plus TARCEVA® group compared with 12.4 months in the placebo plus TARCEVA® group (HR= 0.59; P<0.0001). This PFS benefit was observed across several patient subgroups, and was consistent across Exon 19 and Exon 21 subgroups. Unlike in the FLAURA trial, in the RELAY trial, the PFS in patients with exon 21 mutation was comparable to patients with exon 19 deletions. The PFS for these patients with exon 21 mutation was 19.4 months. Further, the addition of CYRAMZA® to TARCEVA® did not increase the incidence of EGFR T790M mutation. The ORR was 76% in the CYRAMZA® plus TARCEVA® group and 75% in the placebo plus TARCEVA® group, with median DoR of 18.0 months and 11.1 months, respectively. At the time of the final analysis of PFS, the OS data were not mature. The most common adverse reactions in the CYRAMZA® plus TARCEVA® group were infections, hypertension, stomatitis, proteinuria, alopecia, epistaxis, and peripheral edema. The most common laboratory abnormalities were increased ALT and AST as well as cytopenias.
It was concluded that CYRAMZA® plus TARCEVA® demonstrated superior PFS, compared with placebo plus TARCEVA®, in treatment naïve patients with EGFR-mutated metastatic NSCLC. Inhibiting the VEGFR and EGFR pathways together, is an important milestone in the treatment of EGFR-mutated NSCLC, with outcomes comparable to that with third generation TKIs, and furthermore, providing these patients an additional treatment option with TAGRISSO® upon progression.
Ramucirumab plus Erlotinib in Patients with Untreated, EGFR-mutated, Advanced Non-Small-Cell Lung Cancer (RELAY): A Randomised, Double-blind, Placebo-Controlled, Phase 3 trial. Nakagawa K, Garon EB, Seto T, et al. Lancet Oncol. 2019;20:1655-1669.
ENHERTU® Demonstrates Promising Clinical Activity in HER2 Positive Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The HER or erbB family of receptors consist of HER1, HER2, HER3 and HER4. HER2 is a Tyrosine Kinase Receptor expressed on the surface of several tumor types including breast, gastric, lung and colorectal cancers. It is a growth-promoting protein and HER2 overexpression/HER2 gene amplification is often associated with aggressive disease and poor prognosis in certain tumor types. Other HER2 gene alterations such as HER2 mutations, as distinct molecular targets, have been identified in 2-4% of patients with NSCLC, specifically with adenocarcinoma histology. These acquired HER2 gene mutations have been independently associated with cancer cell growth and poor prognosis. There are currently no therapies approved specifically for the treatment HER2 mutant NSCLC, and is therefore an unmet need.
ENHERTU® (Trastuzumab Deruxtecan) is an Antibody-Drug Conjugate (ADC) composed of a humanized monoclonal antibody specifically targeting HER2, with the amino acid sequence similar to HERCEPTIN® (Trastuzumab), attached to a potent cytotoxic Topoisomerase I inhibitor payload by a cleavable tetrapeptide-based linker. ENHERTU® has a favorable pharmacokinetic profile and the tetrapeptide-based linker is stable in the plasma and is selectively cleaved by cathepsins that are up-regulated in tumor cells. Unlike KADCYLA® (ado-Trastuzumab emtansine), which is also an Antibody-Drug Conjugate, ENHERTU® has a higher drug-to-antibody ratio (8 versus 4), the released payload easily crosses the cell membrane with resulting potent cytotoxic effect on neighboring tumor cells regardless of target expression, and the released cytotoxic agent (payload) has a short half-life, minimizing systemic exposure. ENHERTU® is approved in the US for the treatment of adult patients with unresectable or metastatic HER2 positive breast cancer who received two or more prior anti-HER2 based regimens, based on the DESTINY-Breast01 trial.
DESTINY-Lung01 is an ongoing, global, multicenter, open-label, two-cohort, Phase II study, evaluating the safety and efficacy of ENHERTU® in 170 patients with HER2 mutant or HER2 overexpressing (defined as ImmunoHistoChemistry-IHC 3+ or IHC 2+), unresectable and metastatic non-squamous NSCLC. Eligible patients could not have received prior HER2-targeted therapy, with the exception of pan-HER TKIs. Patients were enrolled into 2 cohorts. The first cohort enrolled patients with HER2-expressing tumors as defined by IHC 3+ or 2+ (N = 42). The second cohort included patients whose tumors harbored a HER2 mutation as determined by a local laboratory test (N = 42). Enrolled patients had a median of two prior lines of therapy with majority of patients receiving platinum-based chemotherapy (90.5%), anti-PD-1 or PD-L1 treatment (54.8%) and 19% receiving Docetaxel. Patients received ENHERTU® 4.6 mg/kg every 3 weeks by intravenous infusion. The 42 patients included in the second cohort had a median age of 63 years, 64.3% of patients were female, and 45.2% had CNS metastases. For the majority of patients (90.5%), HER2 mutation was located in the kinase domain. The Primary endpoint was confirmed Objective Response Rate (ORR). Additional endpoints included Disease Control Rate (DCR), Duration of Response (DoR), Progression Free Survival (PFS), and safety. The authors reported data for the cohort with HER2 mutations (second cohort), after a median follow up of 8.0 months.
The ORR was 61.9%, with 2.4% Complete Response, 59.5% Partial Response, and stable disease noted in 28.6% of patients. The Disease Control Rate was 90.5%. The median PFS was 14 months. The median Duration of Response and Overall Survival (OS) had not yet been reached at the time of data cut-off. The most common Grade 3 or higher treatment related Adverse Events were neutropenia and anemia. Confirmed treatment-related Interstitial Lung Disease (ILD) and pneumonitis were noted in approximately 12% of patients and were all Grade 2 and there were no deaths. Nonetheless, ILD is an important identified risk for patients treated with ENHERTU® and requires careful monitoring and management.
It was concluded that ENHERTU® demonstrated promising clinical activity in this interim analysis, with a high Objective Response Rate and durable responses, in a heavily pretreated population of patients with HER2-mutated NSCLC.
Trastuzumab deruxtecan (T-DXd; DS-8201) in patients with HER2-mutated metastatic non-small cell lung cancer (NSCLC): Interim results of DESTINY-Lung01. Smit EF, Nakagawa K, Nagasaka M, et al. J Clin Oncol 38: 2020 (suppl; abstr 9504).
Overall Survival Benefit with Frontline OPDIVO® plus YERVOY® and Limited Chemotherapy in NSCLC
SUMMARY: The FDA on May 26, 2020, approved the combination of OPDIVO® (Nivolumab) plus YERVOY® (Ipilimumab) and 2 cycles of Platinum-doublet chemotherapy as first-line treatment for patients with metastatic or recurrent Non-Small Cell Lung Cancer (NSCLC), with no Epidermal Growth Factor Receptor (EGFR) or Anaplastic Lymphoma Kinase (ALK) genomic tumor aberrations. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the immune system T cells. Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152) and PD-1(Programmed cell Death 1). Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response. OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4.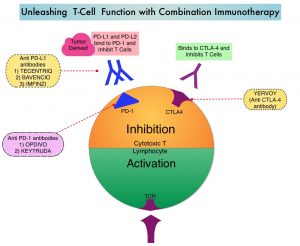
In the CheckMate-227, Part 1, Phase III trial, a combination of OPDIVO® plus YERVOY® significantly improved Overall Survival (OS), Progression Free Survival (PFS), Objective Response Rates (ORR) and Duration of Response, compared to chemotherapy, independent of PD-L1 expression level. The authors in this study hypothesized that a limited course of chemotherapy combined with OPDIVO® plus YERVOY® could provide rapid disease control, while building on the durable Overall Survival benefit seen with dual PD-1 and CTLA-4 inhibition.
CheckMate-9LA is a randomized, open-label, multi-center, Phase III trial which evaluated the benefit of a combination of OPDIVO® plus YERVOY®, and 2 cycles of Platinum-doublet chemotherapy versus Platinum-doublet chemotherapy for 4 cycles followed by optional Pemetrexed maintenance therapy, as a first-line treatment in patients with metastatic or recurrent NSCLC, regardless of PD-L1 status and histology. In this study, 719 adults treatment naïve patients with histologically confirmed Stage IV/recurrent NSCLC, with ECOG Performance Status 0-1, and no known sensitizing EGFR/ALK alterations, were randomly assigned 1:1 to receive OPDIVO® 360 mg every 3 weeks plus YERVOY® 1 mg/kg every 6 weeks and 2 cycles of platinum-doublet chemotherapy (N=361), or 4 cycles of platinum-doublet chemotherapy alone (N=358). Chemotherapy was based on histology. Patients with non-squamous NSCLC in the chemo-only randomized group could receive optional Pemetrexed maintenance treatment. Patients were treated with immunotherapy until disease progression, unacceptable toxicity, or for 2 years. Patients were stratified by PD-L1 status (less than 1% versus 1% or more), sex, and histology (squamous versus non-squamous). Demographics in treatment groups were well balanced. The Primary end point was Overall Survival (OS). Secondary endpoints included Progression Free Survival (PFS), Objective Response Rate (ORR) and efficacy by PD-L1 subgroups.
At a preplanned interim analysis after a minimum follow up 8.1 months, this trial demonstrated a statistically significant benefit in OS for patients treated with OPDIVO® plus YERVOY® and limited chemotherapy, compared to those who received chemotherapy alone. The median OS was 14.1 months versus 10.7 months, respectively (HR=0.69; P=0.0006). With longer follow up at 12.7 months, this OS benefit continued to improve in the immunotherapy plus chemotherapy group, with a median OS of 15.6 months versus 10.9 months, respectively (HR=0.66). The 1-year OS rates were 63% versus 47%. This clinical benefit was consistent across all efficacy measures in key subgroups including by PD-L1 and histology.
The median PFS was 6.8 months in the OPDIVO® plus YERVOY® and chemotherapy group and 5 months in the chemotherapy-only group (HR=0.70; P=0.0001). The ORR was 38% and 25%, respectively (P= .0003). The median response duration was 10 months in the OPDIVO® plus YERVOY® and chemotherapy group, and 5.1 months in the chemotherapy-only group. Grade 3-4 treatment related Adverse Events were reported in 47% of the patients receiving the immunotherapy plus chemotherapy combination versus 38% in the chemotherapy-only group.
It was concluded that CheckMate 9LA met its Primary endpoint of Overall Survival, and OPDIVO® plus YERVOY® with a limited course of chemotherapy should be considered as a new first line treatment option for patients advanced Non Small Cell Lung Cancer.
Nivolumab (NIVO) + ipilimumab (IPI) + 2 cycles of platinum-doublet chemotherapy (chemo) vs 4 cycles chemo as first-line (1L) treatment (tx) for stage IV/recurrent non-small cell lung cancer (NSCLC): CheckMate 9LA. Reck M, Ciuleanu T-E, Dols MC, et al. J Clin Oncol 38: 2020 (suppl; abstr 9501)
Late Breaking Abstract - ASCO 2020: Adjuvant Therapy with TAGRISSO® Improves Survival in Early Stage EGFR-Mutated Non Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R substitution mutation in Exon 21. Approximately 25% of patients with EGFR mutated NSCLC have brain metastases at diagnosis, increasing to approximately 40% within two years of diagnosis. The presence of brain metastases often reduces median survival to less than eight months. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to the most common, acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients.
TAGRISSO® (Osimertinib) is a highly selective third-generation Epidermal Growth Factor Receptor (EGFR) TKI presently approved by the FDA, for the first-line treatment of patients with metastatic NSCLC, whose tumors have Exon 19 deletions or Exon 21 L858R mutations, as well as treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, whose disease has progressed on or after EGFR-TKI therapy. Further, TAGRISSO® has higher CNS penetration and is therefore able to induce responses in 70-90% of patients with brain metastases. Among patients with metastatic, EGFR-mutant NSCLC, first-line treatment with TAGRISSO® significantly improved median Overall Survival, compared with TARCEVA® and IRESSA®, and should therefore be considered the preferred regimen.
Surgical resection is the primary treatment for approximately 30% of patients with NSCLC who present with early Stage (I–IIIA) disease. These patients are often treated with Cisplatin-based adjuvant chemotherapy to decrease the risk of recurrence. Nonetheless, 45-75% of these patients develop recurrent disease. There is therefore an unmet need for this patient population.
ADAURA is a global, double-blind, randomized Phase III study, which assessed the efficacy and safety of TAGRISSO® versus placebo in patients with Stage IB–IIIA EGFR mutated NSCLC, after complete tumor resection and adjuvant chemotherapy, when indicated. In this study, 682 patients with completely resected Stage IB, II, IIIA NSCLC, with or without postoperative adjuvant chemotherapy, were randomly assigned 1:1 to receive either TAGRISSO® 80 mg orally once daily (N=339) or placebo (N=343) once daily, for up to 3 years. Eligible patients had an ECOG Performance Status of 0 or 1, with confirmed EGFR mutations (Exon 19del or L858R). Treatment groups were well balanced and patients were stratified by Stage (IB/II/IIIA), mutation type (Exon 19del/L858R), and race (Asian/non-Asian). The Primary endpoint was Disease Free Survival (DFS) in Stage II–IIIA patients. Secondary endpoints included Overall Survival (OS) and safety. Following Independent Data Monitoring Committee recommendation, the trial was unblinded early, due to efficacy. The authors reported the results from the unplanned interim analysis.
It was noted that in the patients with Stage II/IIIA disease, the DFS had not been reached with TAGRISSO® versus 20.4 months with placebo (HR=0.17; P<0.0001). The 2-year DFS rate in this patient group with TAGRISSO® was 90% versus 44% with placebo. In the overall population, the DFS was still not reached with TAGRISSO® versus 28.1 months with placebo (HR=0.21; P<0.0001). The 2-year DFS rate in the overall population was 89% with TAGRISSO® versus 53% with placebo. The OS data are still early and immature, and the median OS has not yet been reached in either treatment groups. The safety profile was consistent with the known safety profile of TAGRISSO®.
The authors concluded that adjuvant TAGRISSO® is the first targeted agent in a global randomized trial, to show a statistically significant and clinically meaningful improvement in Disease Free Survival, among patients with Stage IB/II/IIIA EGFR mutation-positive NSCLC, and provides an effective new treatment strategy for this patient group.
Osimertinib as adjuvant therapy in patients (pts) with stage IB–IIIA EGFR mutation positive (EGFRm) NSCLC after complete tumor resection: ADAURA. Herbst RS, Tsuboi M, John T, et al. J Clin Oncol 38: 2020 (suppl; abstr LBA5)
FDA Approves RETEVMO® for RET Altered Non Small Cell Lung Cancer and Thyroid Cancers
SUMMARY: The FDA on May 8, 2020, granted accelerated approval to RETEVMO® (Selpercatinib) for patients with metastatic RET fusion-positive Non-Small Cell Lung Cancer (NSCLC), patients with advanced or metastatic RET-mutant Medullary Thyroid Cancer (MTC) who require systemic therapy and those with advanced or metastatic RET fusion-positive thyroid cancer who require systemic therapy and who are RadioActive Iodine (RAI)-refractory. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers.
In addition to the well characterized gene fusions involving ALK and ROS1 in NSCLC, genetic alterations involving other kinases including EGFR, BRAF, RET, NTRK, are all additional established targetable drivers. These genetic alterations are generally mutually exclusive, with no more than one predominant driver in any given cancer. The hallmark of all of these genetic alterations is oncogene addiction, in which cancers are driven primarily, or even exclusively, by aberrant oncogene signaling, and are highly susceptible to small molecule inhibitors.
RET kinase is a transmembrane Receptor Tyrosine Kinase and plays an important role during the development and maintenance of a variety of tissues, including neural and genitourinary tissues. RET signaling activates downstream pathways such as JAK/STAT3 and RAS/RAF/MEK/ERK and leads to cellular proliferation, survival, invasion, and metastasis. Oncogenic alterations to the RET proto-oncogene results in uncontrolled cell growth and enhanced tumor invasiveness. RET alterations include RET rearrangements, leading to RET fusions, and activating point mutations occurring across multiple tumor types. RET fusions have been identified in approximately 2% of NSCLCs, 10-20% of non-medullary thyroid cancers. Activating RET point mutations account for approximately 60% of sporadic Medullary Thyroid Cancers (MTC) and more than 90% of inherited MTCs. Other cancers with documented RET alterations include colorectal, breast, and several hematologic malignancies.
RETEVMO® (Selpercatinib) is a highly selective and potent, oral anti-RET Tyrosine Kinase Inhibitor (TKI) designed to inhibit native RET signaling, as well as anticipated acquired resistance mechanisms. RETEVMO® selectively targets wild-type RET as well as various RET mutants and RET-containing fusion products. Additionally, RETEVMO® inhibits Vascular Endothelial Growth Factor Receptor 1 (VEGFR1), VEGFR3, Fibroblast Growth Factor Receptor 1 (FGFR1), FGFR2, and FGFR3. This results in inhibition of cell growth of tumors that exhibit increased RET activity.
The LIBRETTO-001 is the largest open-label, multicenter, Phase I/II trial in patients with advanced solid tumors, including RET fusion-positive solid tumors, RET-mutant Medullary Thyroid Cancers, and other tumors with RET activation, treated with a RET inhibitor. To investigate the efficacy of RETEVMO®, the trial was conducted in 2 parts: Phase 1 (dose escalation) and Phase II (dose expansion). Patients with advanced cancer were eligible, if they have progressed on or were intolerant to available standard therapies, or no standard or available curative therapy existed, or in the opinion of the Investigator, they would be unlikely to tolerate or derive significant clinical benefit from appropriate standard of care therapy, or they declined standard therapy. A dose of 160 mg BID was the recommended phase 2 dose. Up to about 850 patients with advanced solid tumors harboring a RET gene alteration in tumor and/or blood were enrolled in 6 different Phase 2 cohorts, based on tumor type, RET alteration and prior therapy. Identification of RET gene alterations was prospectively determined in local laboratories using either next generation sequencing, polymerase chain reaction, or fluorescence in situ hybridization. The Phase II portion of the trial had a Primary endpoint of Objective Response Rate (ORR) and Secondary endpoints of Duration of Response, Progression Free Survival (PFS) and safety.
The NSCLC cohort included 105 enrolled patients with RET fusion-positive NSCLC who had received prior platinum-based chemotherapy. Patients had received a median of three prior systemic regimens, 55% had previous treatment with an anti-PD-1/PD-L1 antibody and 48% had previous treatment with at least one multikinase inhibitor. The ORR with RETEVMO® was 64%, and 81% of responding patients had responses lasting 6 months or longer. Efficacy was also evaluated in 39 treatment-naïve patients. The ORR for these patients with RETEVMO® was 85%, and 58% of responding patients had responses lasting 6 months or longer. It is estimated that up to 50% of RET fusion-positive NSCLC patients can have brain metastases, and in the subset of patients with brain metastases in this registrational trial, treatment with RETEVMO® demonstrated a CNS Objective Response Rate of 91%. Median DOR and PFS were not reached at the time of data-cut-off.
In the cohort of advanced or metastatic RET-mutant MTC (N=143), the ORR in patients previously treated with COMETRIQ® (Cabozantinib), CAPRELSA® (Vandetanib), or both (N=55) was 69%, and 76% of responding patients had responses lasting 6 months or longer. Among those patients who had no prior therapy with an approved agent for MTC (N=88), the ORR was 73%, and 61% of responding patients had responses lasting 6 months or longer.
In the cohort of RET fusion-positive thyroid cancer who were RAI-refractory and had received another prior systemic treatment (N=19), the ORR was 79%, and 87% of responders had a response lasting 6 months or longer. Among the patients with RET fusion-positive thyroid cancer who were RAI-refractory and had not received any additional therapy (N=8), the ORR was 100% and 75% of responders had a response lasting 6 months or longer.The most common toxicities included rash, cytopenias, liver function abnormalities, hyperglycemia, hyponatremia, hypocalcemia, increased creatinine and hypertension.
LIBRETTO-001 is the largest trial ever reported in RET-altered cancer patients, and the present FDA approval of RETEVMO® for patients with RET fusions and mutations, across multiple tumor types, represents an important milestone in the Precision Medicine arena.
https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-selpercatinib-lung-and-thyroid-cancers-ret-gene-mutations-or-fusions
FDA Approves CYRAMZA® Plus TARCEVA® for EGFR Mutated NSCLC
SUMMARY: The FDA on May 29, 2020 approved CYRAMZA® (Ramucirumab) in combination with TARCEVA® (Erlotinib) for first-line treatment of metastatic Non-Small Cell Lung Cancer (NSCLC) with Epidermal Growth Factor Receptor (EGFR) Exon 19 deletions or Exon 21 (L858R) mutations. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR mutations and 90% of these mutations are either Exon 19 deletions or L858R substitution mutation in Exon 21. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA®, IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to the most common, acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients. Previously published data from the Phase III FLAURA study showed that first-line treatment with third generation TKI, TAGRISSO® (Osimertinib), was superior to first-line treatment with other first and second generation TKI’s, in patients with EGFR-mutated NSCLC. However, widespread use of TAGRISSO® has led to acquired resistance. Novel treatment approaches combining TKI’s with other targeted therapies are therefore needed.
CYRAMZA® is a recombinant human monoclonal IgG1 antibody that binds to the human Vascular Endothelial Growth Factor Receptor- 2 (VEGFR-2), preventing the interaction of VEGFR-2 with its ligands. TARCEVA® is a first generation EGFR TKI. Preclinical and clinical data strongly support dual blockade of the EGFR and VEGF pathways in EGFR-mutated metastatic NSCLC.
RELAY is an International, double-blind, Phase III trial, which included 449 eligible patients who had Stage IV NSCLC, with an EGFR Exon 19 deletion (ex19del) or Exon 21 substitution (L858R) mutation, and with no CNS metastases. Enrolled patients were randomly assigned in a 1:1 ratio to receive TARCEVA® 150 mg orally daily plus CYRAMZA® 10 mg/kg IV once every 2 weeks (N=224) or TARCEVA® plus a matching placebo (N=225). Patients were stratified by sex, EGFR mutation type, and EGFR testing methodology. The Primary endpoint was Progression Free Survival (PFS) and key Secondary endpoints included Safety, Overall Response Rate (ORR), Duration of Response, and Overall Survival (OS).
At a median follow up of 20.7 months, PFS was significantly longer in the TARCEVA® plus CYRAMZA® group compared to TARCEVA® plus placebo group (19.4 months versus 12.4 months respectively; HR=0.59; P<0.0001). This benefit was observed regardless of tumor type, and was consistent across Exon 19 and Exon 21 subgroups. The ORR was similar between the CYRAMZA® and placebo groups (76% versus 75%), but the median Duration of Response was longer in the CYRAMZA® group, compared with the placebo group (18 months versus 11 months). The OS data were not mature at the time of final PFS analysis and the median time to the second disease progression (PFS2) was not yet reached. However, interim results indicated that PFS2 was longer in the CYRAMZA® group compared to the placebo group (HR = 0.69) suggesting that PFS benefits with CYRAMZA® were preserved beyond first progression, indicating that possibility of OS benefit. Upon progression, T790M resistance mutations were detected in 43% of patients who received CYRAMZA®, and in 47% of patients who received placebo. The most common adverse events in the TARCEVA® plus CYRAMZA® combination included infections, stomatitis, hypertension, proteinuria, alopecia, epistaxis and peripheral edema.
It was concluded that TARCEVA® plus CYRAMZA® demonstrated superior PFS compared with TARCEVA® plus placebo, in treatment naïve patients with EGFR-mutated metastatic NSCLC. The combination of TARCEVA® plus CYRAMZA® will be a new additional treatment option for this patient group.
Ramucirumab plus Erlotinib in Patients with Untreated, EGFR-mutated, Advanced Non-Small-Cell Lung Cancer (RELAY): A Randomised, Double-blind, Placebo-Controlled, Phase 3 trial. Nakagawa K, Garon EB, Seto T, et al. Lancet Oncol. 2019;20:1655-1669.
FDA Approves ALUNBRIG® for First Line Treatment of ALK Positive Non Small Cell Lung Cancer
SUMMARY: The FDA on May 22, 2020 approved approved ALUNBRIG® (Brigatinib) for the first-line treatment of patients with ALK-positive metastatic Non Small Cell Lung Cancer (NSCLC), as detected by an FDA-approved test. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The discovery of rearrangements of the Anaplastic Lymphoma Kinase (ALK) gene in some patients with advanced NSCLC and adenocarcinoma histology, led to the development of agents such as XALKORI® (Crizotinib), ZYKADIA® (Ceritinib), ALECENSA® (Alectinib) and ALUNBRIG® (Brigatinib), with promising results. It has become clear that appropriate, molecularly targeted therapy for tumors with a molecular abnormality, results in the best outcomes. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8% as well as other mutations in BRAF, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive, and the presence of two simultaneous mutations, are rare.
The new approval for ALUNBRIG® was based on results from the Phase III ALTA 1L (ALK in Lung Cancer Trial of BrigAtinib in 1st Line) trial, which is a global, ongoing, randomized, open-label, comparative, multicenter study, in which investigators compared the efficacy and safety of ALUNBRIG® with XALKORI® (Crizotinib) in 275 patients with Stage IIIB/IV ALK positive, locally advanced or metastatic NSCLC, who have not received prior treatment with an ALK inhibitor, but may have received 1 prior regimen of chemotherapy or no chemotherapy in the advanced setting. Patients were randomized 1:1 to receive either ALUNBRIG® 180 mg orally once daily (N=137), with a 7-day lead-in period at 90 mg, or XALKORI® 250 mg orally twice daily (N=138). Crossover from the XALKORI® arm to receive ALUNBRIG® was permitted at BICR (Blinded Independent Review Committee)-assessed Progression Free Survival (PFS). The median age was 59 years, and 55% of patients were female. Twenty-nine percent had brain metastases at baseline with comparable pre-enrollment central nervous system (CNS) radiotherapy rates among both cohorts. Overall, 27% of patients had prior chemotherapy in the locally advanced or metastatic setting. The Primary endpoint was BIRC assessed PFS and Secondary endpoints included Objective Response Rate (ORR), Intracranial ORR, Intracranial PFS, Overall Survival (OS), safety, and tolerability.
At a median follow up of 25 months, it was noted that ALUNBRIG® reduced the risk of disease progression or death by 51% compared with XALKORI® (HR=0.49; P=0.0007), with a median PFS of 24 months as assessed by a BIRC versus 11 months for XALKORI®. The confirmed ORR as assessed by BIRC was 74% with ALUNBRIG® and 62% for XALKORI®. The median duration of response (DOR) was not reached, and 13.8 months with ALUNBRIG® and XALKORI®, respectively.
After more than two years of follow-up, ALUNBRIG® demonstrated superiority over XALKORI®, with significant anti-tumor activity observed, especially in patients with baseline brain metastases. The confirmed intracranial ORR for patients with measurable brain metastases at baseline, treated with ALUNBRIG® was 78% versus 26% for patients treated with XALKORI®. The median intracranial Duration of Response in confirmed responders with measurable brain metastases at baseline was Not Reached with ALUNBRIG® and 9.2 months with XALKORI®, respectively. The median intracranial PFS was 24 months with ALUNBRIG®, compared with 5.6 months for XALKORI®. ALUNBRIG® reduced the risk of intracranial disease progression or death by 69% in patients who had brain metastases at baseline (HR=0.31).
Additionally, patients in the ALUNBRIG® group also experienced significant improvements in Health-Related Quality of Life, with delay in the median time to worsening in Global Health Score by 27 months versus 8 months with XALKORI®, as well as delay in the time to worsening and prolonged duration of improvement in fatigue, nausea and vomiting, appetite loss, and emotional and social functioning. Further, the duration of improvement in QoL with ALUNBRIG® was Not Reached versus 12 months with XALKORI®.
It was concluded that ALUNBRIG® demonstrated a statistically and clinically significant improvement in Progression Free Survival when compared to XALKORI® in ALK inhibitor-naïve, ALK positive NSCLC, with superior efficacy especially among those with brain metastases at baseline.
Brigatinib vs crizotinib in patients with ALK inhibitor-naive advanced ALK+ NSCLC: Updated results from the phase III ALTA-1L trial. Camidge R, Kim HR, Ahn M, et al. Presented at the 2019 ESMO Asia Congress, November 23, 2019.
FDA Approves Chemotherapy-Free First Line Immunotherapy Combination in Advanced NSCLC
SUMMARY: The FDA on May 15, 2020, approved OPDIVO® (Nivolumab) in combination with YERVOY® (Ipilimumab), as first-line treatment for patients with metastatic Non-Small Cell Lung Cancer (NSCLC), whose tumors express PD-L1(1% or more), as determined by an FDA-approved test, with no Epidermal Growth Factor Receptor (EGFR) or Anaplastic Lymphoma Kinase (ALK) genomic tumor aberrations. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the immune system T cells. Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152) and PD-1(Programmed cell Death 1). Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response. OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. OPDIVO® is presently approved for treatment of patients with metastatic NSCLC and progression on or after Platinum-based chemotherapy. YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4.
The present FDA approval was based on CheckMate-227, which is an open-label, multi-part, global, Phase III trial in which OPDIVO® based regimens were compared with Platinum-doublet chemotherapy in patients with first line advanced NSCLC, across non-squamous and squamous tumor histologies. In Part 1 of this trial, there were 2 cohorts- Part 1a in which OPDIVO® plus low dose YERVOY® (N=396) or OPDIVO® monotherapy (N=396) was compared with chemotherapy alone (N=397), in patients whose tumors expressed PD-L1 expression of 1% or more and Part 1b in which OPDIVO® plus low dose YERVOY® (N=187) or OPDIVO® plus chemotherapy (N=177) was compared with chemotherapy alone (N=186), in patients whose tumors did not express PD-L1 (less than 1%). (In Part 2 of this trial, OPDIVO® plus chemotherapy was compared with chemotherapy alone, regardless of PD-L1 expression. Part 2 did not meet its Primary endpoint for Overall Survival for OPDIVO® plus chemotherapy versus chemotherapy alone, in patients with non-squamous NSCLC, and is published elsewhere). It should be noted that when this trial was launched, chemotherapy along with immunotherapy or immunotherapy alone was not approved for the front-line treatment of NSCLC. Therefore, dual immunotherapy combination was not compared with current standards of care such as chemotherapy plus immunotherapy.
OPDIVO® was administered at 3 mg/kg every 2 weeks, and in the combination arm, YERVOY® was administered at 1 mg/kg every 6 weeks. When administered with chemotherapy, OPDIVO® was administered at 360 mg every 3 weeks. Patients were stratified by histology, and treatment was administered until disease progression, unacceptable toxicity, or administered for 2 years for immunotherapy. There were two Co-primary endpoints in Part 1 for OPDIVO® plus YERVOY® versus chemotherapy: Overall survival (OS) in patients whose tumors express PD-L1 (assessed in patients enrolled in Part 1a) and Progression Free Survival (PFS) in patients with TMB of 10 mut/Mb or more, across the PD-L1 spectrum (assessed in patients enrolled across Parts 1a and 1b). The minimum follow up for the Primary endpoint was 29 months. Both Part 1a and Part 1b groups met their Primary endpoints.
In the Part 1a cohort with PD-L1 expression of 1% or more, the Overall Survival was significantly longer with OPDIVO® plus YERVOY®, compared to chemotherapy. The median Overall Survival was 17.1 months in the Immunotherapy combination group compared to 14.9 months in the chemotherapy group (HR=0.79; P=0.007), with a 2-year OS rates of 40.0% and 32.8%, respectively. Progression Free Survival, Objective Response Rates and Duration of Response were all greater with OPDIVO® plus YERVOY® combination, compared to chemotherapy. The median Progression Free Survival (PFS) was 5.1 months in the OPDIVO® plus YERVOY® group and 5.6 months in the platinum-doublet chemotherapy group (HR=0.82). Confirmed Overall Response Rate (ORR) was 36% and 30% respectively. Median Duration of Response was 23.2 months in the OPDIVO® plus YERVOY® group and 6.2 months in the platinum-doublet chemotherapy group. In the Part 1b cohort with PD-L1 expression of less than 1%, Overall Survival benefit was again observed with the OPDIVO® plus YERVOY® combination, compared with chemotherapy, with a median duration of 17.2 months with OPDIVO® plus YERVOY® and 12.2 months with chemotherapy. Among all the patients in the trial, the median duration of OS was 17.1 months with OPDIVO® plus YERVOY® and 13.9 months with chemotherapy. Grade 3 and 4 treatment-related Adverse Events across all patients was 33% in those treated with OPDIVO® plus YERVOY® combination and 36% with chemotherapy.
It was concluded that first-line treatment of patients with advanced NSCLC, with a combination of two immunotherapy drugs, improves Overall Survival, compared to chemotherapy, independent of the PD-L1 expression level, and offers a chemotherapy-free first line treatment option for a subset of NSCLC patients, leaving chemotherapy for later lines of therapy.
Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. N Engl J Med. 2019;381:2020-2031.
FDA Approves TABRECTA® for Metastatic Non-Small Cell Lung Cancer
SUMMARY: The FDA on May 6, 2020, granted accelerated approval to TABRECTA® (Capmatinib) for adult patients with metastatic Non-Small Cell Lung Cancer (NSCLC), whose tumors have a mutation that leads to Mesenchymal-Epithelial Transition (MET) exon 14 skipping, as detected by an FDA-approved test. The FDA also approved the FoundationOne CDx assay (Foundation Medicine, Inc.) as a companion diagnostic for TABRECTA®.
MET is a widely expressed Receptor Tyrosine Kinase and plays a pivotal role in cell growth, proliferation and survival. The MET gene encodes for a protein known as the Hepatocyte Growth Factor (HGF) Receptor. Upon binding by Hepatocyte Growth Factor (HGF), the HGF Receptor is activated, with resulting activation of the downstream RAS/RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways, thereby serving different important biological functions. Alterations in the MET gene leading to abnormal MET signaling, has been identified in different types of cancers including thyroid, lung, breast, liver, colon, kidney, ovary and gastric carcinoma.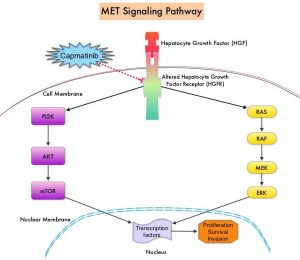
Two key MET alterations include MET exon 14 skipping mutations and MET amplification. MET exon 14 skipping mutations occur in approximately 5% of NSCLC patients with enrichment in sarcomatoid lung cancers (22%). MET exon 14 skipping mutation is a recognized oncogenic driver and is a molecular genetic abnormality indicating the presence of a splice site mutation that results in a loss of transcription of exon 14 of the MET gene. Most exon 14 mutations occur in never-smokers and is seen in both squamous and adenocarcinoma histology. Patients whose cancers have MET exon 14 skipping generally have very high response rates to MET inhibitors and molecular testing for MET exon 14 skipping should therefore be performed on all lung cancers, because this is a targetable alteration. MET amplification has been more commonly seen in smokers, and responses in patients with MET-amplified tumors might be more variable and dependent on level of amplification, with higher responses noted in tumors with more than 5-6 fold amplification. Tumors with MET exon 14 skipping mutations usually do not harbor activating mutations in EGFR, KRAS, or BRAF or concurrent ALK, ROS1 or RET translocations. However, it appears that cMET exon 14 skipping is not mutually exclusive with cMET amplification.
TABRECTA® (Capmatinib) is a highly potent and selective, reversible inhibitor of MET tyrosine kinase. The present FDA approval was based on the primary findings from the Phase II GEOMETRY mono-1 trial, which is a non-randomized, open-label, multi-cohort, Phase II study, conducted to evaluate the efficacy and safety of single-agent TABRECTA® in adult patients with EGFR wild-type, ALK-negative, metastatic NSCLC, whose tumors have a mutation that leads to MET exon 14 skipping (METex14), as detected by an RNA-based RT-PCR. This study enrolled 97 patients with metastatic NSCLC and confirmed MET exon 14 skipping mutations, 69 of whom were previously treated and, 28 of whom, were treatment naive. The patients received TABRECTA® at 400 mg orally twice daily until disease progression or unacceptable toxicity. The median patient age was 71 years and all NSCLC histologies including sarcomatoid/carcinosarcoma were included. Majority of the patients (75%) were white and 24% were Asian. Previous treatments included immunotherapy (28%) and chemotherapy (94%), and 23% of patients received 2 prior lines of therapy. The main efficacy outcome was Overall Response Rate (ORR) and additional efficacy outcomes included Duration of Response, Time to Response, Disease Control Rate, Progression Free Survival (PFS) and Safety. Thirteen patients (N=13) in this study had brain metastases at baseline.
Among the treatment-naïve patients group, the ORR was 68% with a median Duration of Response of 12.6 months and the percentage of patients with responses for 12 months or longer was 47%. The Disease Control Rate (Complete Response plus Partial Response plus Stable Disease) was 96.4%.
Among the previously treated patients, the ORR was 41%, with a median Duration of Response of 9.7 months and the percentage of patients with responses for 12 months or longer was 32%. The Disease Control Rate was 78.3%. Among those with brain metastases at baseline, 54% had an intracranial response with TABRECTA® with 31% showing complete resolution, 23% showing partial resolution, and the intracranial Disease Control Rate was 92%. The most common adverse events (occurring in at least 20% of patients) were peripheral edema, nausea, fatigue, vomiting, dyspnea, and decreased appetite. TABRECTA® can also cause Interstitial Lung Disease, hepatotoxicity and photosensitivity.
It was concluded that TABRECTA® is a new treatment option for patients with MET exon 14 skipping- mutated advanced NSCLC, regardless of the line of therapy, with deep and durable responses, manageable toxicity profile, and is the first and only FDA approved treatment for this patient group.
Capmatinib (INC280) in METex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. Wolf J, Seto T, Han J, et al. J Clin Oncol. 2019;37(suppl; abstr 9004).
NELSON Trial Confirms that Low-Dose CT Screening Reduces Lung Cancer Mortality
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
In the National Lung Screening Trial (NLST) with Low Dose CT (LDCT) screening for lung cancer, there was a 20% reduction in mortality. Following the publication of the results of NLST and NCCN issued guideline in 2011, the United States Preventive Services Task Force (USPSTF) recommended Lung Cancer screening with Low Dose CT scan in high risk patients. CMS in 2015 determined that there was sufficient evidence to reimburse for this preventive service.
Despite the evidence and recommendation along with supportive public policies, screening with LDCT has not been adequately implemented in the US healthcare system. Low Health Care Provider knowledge of the Lung Cancer Screening (LCS) guidelines represents a potential barrier to implementation. Additionally, despite the unequivocal findings from the NLST, several countries have not adopted this guideline due to early publication of inconclusive results from a number of smaller trials in Europe. Further, there are limited data from randomized trials regarding whether nodule volume-based, low-dose CT screening can reduce lung cancer mortality among male former and current smokers.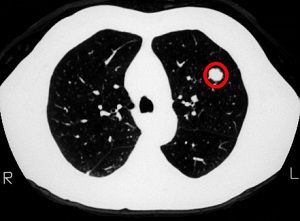
The Dutch-Belgian lung cancer screening trial, NELSON (Nederlands–Leuvens Longkanker Screenings Onderzoek ) is a population-based, randomized, controlled trial initiated in 2000. The goal of the study was to show a 25% or more reduction in lung cancer mortality, in high-risk male participants at 10 years of follow-up, utilizing volume-based, low-dose CT lung cancer screening. The authors in this publication reported the incidence of lung cancer, associated mortality, and the performance of the four rounds of low-dose CT screening for lung cancer in the this trial, among male participants (main analysis) and female participants (subgroup analyses).
In this study, a total of 13,195 men aged 50-74 years were randomly assigned to undergo CT screening at baseline, year 1, year 3, and year 5.5 (N = 6,583) or no screening (N=6,612). At the time of initiation of this trial, only a small number of women were eligible, as smoking was much less prevalent and intense among women, than among men. Because of the relevance and importance of the inclusion of women in this study, high-risk women were later allowed to participate (N=2594). Participants were current or former smokers who had quit 10 or fewer years ago, who had smoked more than 15 cigarettes a day for more than 25 years, or more than 10 cigarettes a day for more than 30 years. About 45% of the male participants were former smokers.
At 10 years of follow up, lung cancer mortality among men was 24% lower in the screening group than in the control group (Rate Ratio=0.76; P=0.01), and 33% lower among women (Rate Ratio=0.67). The CT screening compliance among men was 90% and approximately 9.2% of the screened participants underwent at least one additional CT scan due to an indeterminate screening test. Screening-detected lung cancers were substantially more often diagnosed in Stage IA or IB (58.6%) and were Adenocarcinomas (52.0% in the screening group and 43.8% in the control group).
It was concluded that in this trial involving high-risk individuals, lung cancer mortality was significantly lower among those who underwent CT screening with determination of nodule volume, than among those who underwent no screening. Adherence to CT screening was very high, with low rates of follow up procedures for results suggestive of lung cancer. Reduced Lung-Cancer Mortality with Volume CT Screening in a Randomized Trial. de Koning H.J., van der Aalst C.M., de Jong P.A., et al. N Engl J Med 2020;382:503-513
Lung Immune Prognostic Index (LIPI) is an Important Prognostic Biomarker for Patients with Advanced Non Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immunotherapy with PD-1/PD-L1 (Programmed Death-1/Programmed Death-Ligand 1) inhibitors, also called Immune Checkpoint Inhibitors (ICIs), has changed the treatment paradigm for patients with advanced NSCLC. In previously treated patients with NSCLC, the Overall Response Rates (ORR) with single agent Immune Checkpoint Inhibitors (ICIs) range from 14-20%, with median Overall Survival (OS) of 10 to 12 months. In those with PD-L1 expression of 50% or more by ImmunoHistoChemical (IHC) analysis, the ORR can reach up to 30% with a median OS of 20 months. However, in patients with negative or weak PD-L1 expression (1%-49% positive tumor cells), who account for approximately two thirds of the NSCLC population, the response rates range from 8-19% with a median OS slightly below 10 months. Even among those with tumors expressing PD-L1 expression of 50% or more, not all patients benefit from Immunotherapy with ICIs. Therefore identifying biomarkers for patients likely to respond to ICI therapy, and predicting resistance is important and relevant in selecting the appropriate patients for treatment with ICIs.
There is growing evidence on the role of inflammation in cancer biology and systemic inflammatory response may have prognostic significance in different cancer types. Inflammatory process in various cancers imparts immunoresistance to ICIs, by activating oncogenic signaling pathways, there by promoting cancer growth and dissemination, with resulting poor outcomes. Derived Neutrophil-to-Lymphocyte ratio (dNLR) and serum Lactate DeHydrogenase (LDH) level have been investigated as potential inflammatory biomarkers in patients with cancer. The dNLR is calculated using a formula dNLR= Absolute Neutrophil Count/(White Blood Count minus Absolute Neutrophil Count). These ratios are simple and easy to calculate from routine blood tests. Both these biomarkers have been correlated with Immune Checkpoint Inhibitor outcomes, in patients with melanoma. In two large studies involving patients with advanced melanoma treated with Ipilimumab and Pembrolizumab, dNLR of 3 or more and LDH of at least 2.5 times Upper Limit of Normal (ULN), reflected a pro-inflammatory status and resulted in poor outcomes.
Based on this important finding in malignant melanoma, Mezquita L and colleagues (JAMA Oncol. 2018;4:351-357) conducted a multicenter, retrospective study involving 466 patients treated with ICIs, to determine whether combining the two factors - pretreatment dNLR and LDH (Lung Immune Prognostic Index-LIPI), was associated with resistance to ICIs in patients with advanced NSCLC. In this study, LIPI was developed on the basis of dNLR (derived Neutrophil-to-Lymphocyte Ratio) of greater than 3 and LDH greater than Upper Limit of Normal (ULN). LIPI was used to stratify patients with NSCLC into 3 groups (Good= 0 factors; Intermediate= 1 of 2 factors, Poor= 2 factors). The authors based on this study concluded that pretreatment LIPI, combining derived Neutrophil-to-Lymphocyte ratio (dNLR) greater than 3 and serum LDH level greater than Upper Limit of Normal, correlated with worse outcomes for Immune Checkpoint Inhibitors (ICIs).
To determine whether LIPI score provides prognostic information for patients with metastatic NSCLC, the authors in this publication performed an exploratory retrospective analysis of the LIPI on pooled clinical trial data from 11 randomized multinational studies (5 ICI trials and 6 targeted therapy trials), and in the final analysis included 3987 patients treated with ICIs, targeted therapy, or cytotoxic chemotherapy, between January 1, 2013, and December 31, 2017. In the 5 ICI trials (N = 2440), 1368 patients received ICIs and 1072 received cytotoxic chemotherapy. In the 6 targeted therapy trials (N = 1547), 53% of EGFR mutant and 47.1% of ALK positive patients received targeted therapy 32.0% of EGFR mutant and 68% of ALK positive patients received cytotoxic chemotherapy. Baseline demographics and disease characteristics were relatively balanced between groups. Lung Immune Prognostic Index (LIPI) scores were calculated based on the dNLR and the LDH level, as mentioned elsewhere in this document.
For patients receiving ICIs, a good LIPI score was associated with longer Overall Survival (OS) compared with a poor LIPI score, with an estimated median survival of 15.6 versus 4.5 months (HR=0.34). A similar prognostic association was observed for patients who received cytotoxic chemotherapy, with patients having a good LIPI score having a longer survival than patients with a poor score, with an estimated median survival of 10.4 versus 5.3 months (HR=0.49). Similar associations were also noted between good LIPI scores and longer Progression Free Survival (PFS). As expected, PD-L1 expression of 1% or more, as well as higher albumin levels was independently associated with improved outcomes.
Among patients with tumors harboring either ALK alterations or EGFR-activating mutations who received targeted therapy, those with a good LIPI score had an estimated median survival of 46.5 months compared with 16.6 months for those with a poor score (HR=0.28). A similar prognostic association was observed in this patient group receiving cytotoxic chemotherapy, with patients having a good LIPI score experiencing a longer survival than patients with a poor score (estimated median survival of 33.4 months versus 17.1 months (HR=0.41). Further, similar associations between LIPI score and PFS were observed. For patients enrolled in these studies, regardless of receiving targeted therapy or cytotoxic chemotherapy, multivariable analysis consistently showed that LIPI score was independently associated with OS and PFS.
It was concluded from this analysis that pretreatment LIPI risk score may be an important prognostic biomarker, irrespective of pharmacologic class of treatment, for patients with metastatic NSCLC. Prognostic Value of the Lung Immune Prognostic Index for Patients Treated for Metastatic Non–Small Cell Lung Cancer. Kazandjian D, Gong Y, Keegan P, et al. JAMA Oncol. 2019;5:1481-1485.
FDA Approves Frontline TECENTRIQ® with Carboplatin and nab-Paclitaxel for Metastatic Non-Squamous NSCLC
SUMMARY: The FDA on December 3, 2019 approved TECENTRIQ® (Atezolizumab) in combination with nab-Paclitaxel and Carboplatin for the first-line treatment of adult patients with metastatic non-squamous Non-Small Cell Lung Cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019, about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the T cells of the immune system. Immuno-Oncology (IO) therapies unleash the T cells by blocking the Immune checkpoint proteins, thereby resulting in T cell proliferation, activation and a therapeutic response. Immunotherapy with PD-1 (Programmed cell Death 1) and PD-L1 (Programmed cell Death Ligand 1) inhibitors have demonstrated a clear survival benefit both as a single agent or in combination, compared with standard chemotherapy, in both treatment-naive and previously treated patients for advanced NSCLC. It is now standard therapy for patients with lung cancer. TECENTRIQ® is an anti PD-L1 monoclonal antibody, designed to directly bind to PD-L1, expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors and thus enabling the activation of T cells. 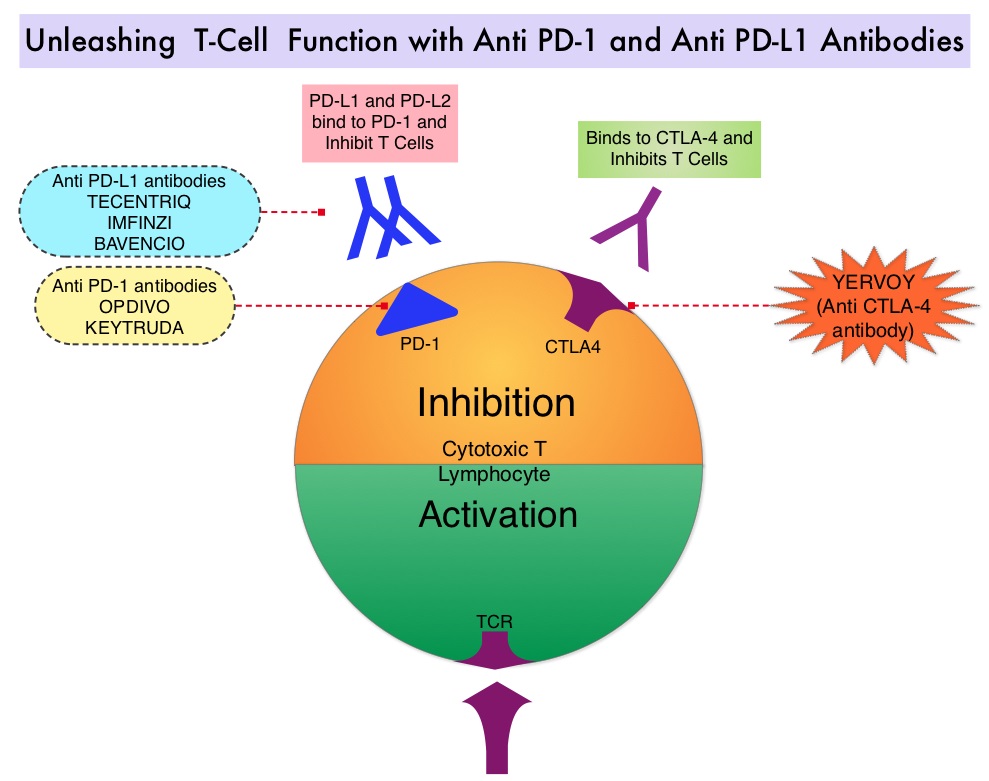
IMpower 130 is an international, multicentre, open-label, randomized, Phase III study, evaluating the efficacy and safety of TECENTRIQ® in combination with Carboplatin and nab-Paclitaxel versus chemotherapy (Carboplatin and nab-Paclitaxel) alone, for chemotherapy-naïve patients with Stage IV non-squamous NSCLC. This study enrolled 724 patients who were randomly assigned 2:1 to receive TECENTRIQ® 1200 mg IV on Day 1, along with Carboplatin AUC 6 on Day 1 and nab-Paclitaxel 100 mg/m2 IV, on days 1, 8 and 15 of each 21-day cycle, for 4 or 6 cycles followed by maintenance TECENTRIQ®, or Carboplatin and nab-Paclitaxel alone (control group), followed by Best Supportive Care during the maintenance treatment phase or Switch maintenance to Pemetrexed every 3 weeks. Stratification factors included gender, baseline liver metastases, and PD-L1 expression. The co-Primary endpoints were investigator-assessed Progression Free Survival and Overall Survival in the intention-to-treat EGFR and ALK wild-type population. The median follow up in the population studied was 19 months.
There was a significant improvement in median Overall Survival in the TECENTRIQ® plus chemotherapy group at 18.6 months compared to 13.9 months in the chemotherapy alone group (HR=0.79; P=0.033), as well as improvement in the median Progression Free Survival (7 months versus 5.5 months, respectively, HR=0.64; P<0.0001). The most common adverse reactions reported in 20% or more of patients in the TECENTRIQ® and chemotherapy group were fatigue/asthenia, nausea, alopecia, constipation, diarrhea, and decreased appetite.
It was concluded that first-line TECENTRIQ® in combination with chemotherapy significantly improved Overall Survival and Progression Free Survival, compared to chemotherapy alone, in patients with advanced non-squamous NSCLC without ALK or EGFR mutations. This IO-chemotherapy combination is the second FDA approval for this patient population. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. West H, McCleod M, Hussein M, et al. Lancet Oncol. 2019;20:924-937
Late Breaking Abstract - ESMO 2019: First-Line Treatment with TAGRISSO® Improves Overall Survival in EGFR-Positive NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019, about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R substitution mutation in Exon 21. Approximately 25% of patients with EGFR mutated NSCLC have brain metastases at diagnosis, increasing to approximately 40% within two years of diagnosis. The presence of brain metastases often reduces median survival to less than eight months. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to the most common, acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients.
TAGRISSO® (Osimertinib) is a highly selective third-generation Epidermal Growth Factor Receptor (EGFR) TKI presently approved by the FDA, for the first-line treatment of patients with metastatic NSCLC, whose tumors have Exon 19 deletions or Exon 21 L858R mutations, as well as treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, whose disease has progressed on or after EGFR-TKI therapy. Further, TAGRISSO® has higher CNS penetration and is therefore able to induce responses in 70-90% of patients with brain metastases.
FLAURA, which is a randomized, double blind, Phase III clinical trial, was conducted to compare the efficacy and safety of first line TAGRISSO® to TARCEVA® or IRESSA® (the latter two are considered standard first line therapies), in NSCLC patients with sensitizing EGFR mutations. This study randomized 556 advanced NSCLC treatment naïve patients, with EGFR Exon 19 Deletions or Exon 21 (L858R) substitution mutations, in a 1:1 ratio, to TAGRISSO® 80 mg orally once daily (N=279) or Standard of Care EGFR-TKI, IRESSA® 250 mg or TARCEVA® 150 mg, orally once daily (N=277). Patients were stratified by mutation status (Exon 19 vs 21 mutations) and race (Asian vs non-Asian). Patients with CNS metastases who were neurologically stable, were allowed in this study. The Primary endpoint was Progression Free Survival (PFS) and Overall Survival (OS) was a Secondary endpoint.
The median PFS was 18.9 months with TAGRISSO®, compared to 10.2 months for the standard therapy (HR=0.46; P<0.001), suggesting a 54% reduction in the risk of disease progression, compared with Standard of Care. TAGRISSO® extended the median Time To Progression by about 9 months. This PFS benefit was consistent across all subgroups of patients, including those with and without CNS metastases at study entry. The Objective Response Rate (ORR) with TAGRISSO® was 80% compared with 76% for TARCEVA® and IRESSA®. The median Duration of Response with TAGRISSO® was 17.2 months versus 8.5 months in the comparator arm.
The authors now reported the Overall Survival (OS) results from the Phase III FLAURA trial. Approximately, 25% of patients in the comparator group had crossed over to the TAGRISSO® group upon disease progression. Despite this crossover, the OS was significantly prolonged with TAGRISSO® with a median OS of 38.6 months compared with 31.8 months for the comparator arm (HR=0.799; P=0.046), representing a 20% reduction in the risk of death with TAGRISSO®. At the time of final data cutoff for the study, about 54% of patients remained alive at 3 years in the TAGRISSO® group compared with 44% in the comparator group, and 28% of patients enrolled in the trial were still receiving TAGRISSO® at three years versus 9% on either TARCEVA® or IRESSA®. Treatment with TAGRISSO® also resulted in a statistically significant and clinically meaningful 52% reduction in the risk of CNS disease progression or death, compared to the comparator arm (HR=0.48; P=0.014). Fewer patients in the TAGRISSO® group experienced Grade 3 or more Adverse Events compared to the comparator arm. Approximately 30% of patients in both treatment groups did not receive subsequent therapy after progression.
It was concluded that among patients with metastatic, EGFR-mutant NSCLC, first-line treatment with TAGRISSO®, significantly improved median Overall Survival, compared with TARCEVA® and IRESSA®, and should therefore be considered the preferred regimen. Osimertinib vs comparator EGFR-TKI as first-line treatment for EGFRm advanced NSCLC (FLAURA): Final overall survival analysis. Ramalingam SS, Gray JE, Ohe Y, et al. Presented at 2019 ESMO Congress, Barcelona, Spain, September 28 to October 1, 2019. Abstract LBA5_PR.
Late breaking Abstract - ESMO 2019: Chemotherapy-Free First Line Immunotherapy Combination Improves Overall Survival in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019, about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions by switching off the T cells of the immune system.
Immune checkpoint proteins/receptors include CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152) and PD-1(Programmed cell Death 1). Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response. OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. OPDIVO® is presently approved for treatment of patients with metastatic NSCLC and progression on or after Platinum-based chemotherapy. YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4).
CheckMate-227 is an open-label, multi-part, global, Phase III trial in which OPDIVO® based regimens were compared with Platinum-doublet chemotherapy in patients with first line advanced NSCLC, across non-squamous and squamous tumor histologies. In Part 1 of this trial, there were 2 cohorts- Part 1a in which OPDIVO® plus low dose YERVOY® (N=396) or OPDIVO® monotherapy (N=396) was compared with chemotherapy alone (N=397), in patients whose tumors expressed PD-L1 expression of 1% or more and Part 1b in which OPDIVO® plus low dose YERVOY® (N=187) or OPDIVO® plus chemotherapy (N=177) was compared with chemotherapy alone (N=186), in patients whose tumors did not express PD-L1 (less than 1%). (In Part 2 of this trial, OPDIVO® plus chemotherapy was compared with chemotherapy alone, regardless of PD-L1 expression. Part 2 did not meet its Primary endpoint for Overall Survival for OPDIVO® plus chemotherapy versus chemotherapy alone, in patients with non-squamous NSCLC, and is published elsewhere). It should be noted that when this trial was launched, chemotherapy along with immunotherapy or immunotherapy alone was not approved for the front-line treatment of NSCLC. Therefore, dual immunotherapy combination was not compared with current standards of care such as chemotherapy plus immunotherapy.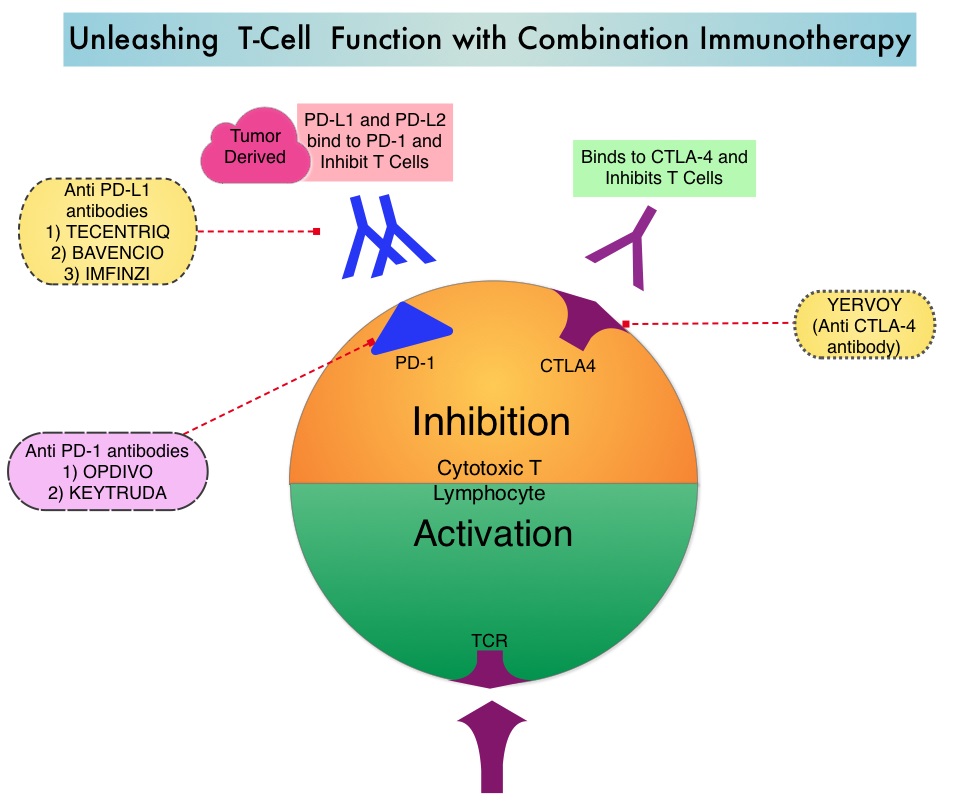
OPDIVO® was administered at 3 mg/kg every 2 weeks, and in the combination arm, YERVOY® was administered at 1 mg/kg every 6 weeks. When administered with chemotherapy, OPDIVO® was administered at 360 mg every 3 weeks. Patients were stratified by histology, and treatment was administered until disease progression, unacceptable toxicity, or for 2 years, for immunotherapy. There were two Co-primary endpoints in Part 1 for OPDIVO® plus YERVOY® versus chemotherapy: Overall survival (OS) in patients whose tumors express PD-L1 (assessed in patients enrolled in Part 1a) and Progression Free Survival (PFS) in patients with TMB of 10 mut/Mb or more, across the PD-L1 spectrum (assessed in patients enrolled across Parts 1a and 1b). The minimum follow up for the Primary endpoint was 29 months.
Both Part 1a and Part 1b groups met their Primary endpoints. In the Part 1a cohort with PD-L1 expression of 1% or more, the Overall Survival was significantly longer with OPDIVO® plus YERVOY®, compared to chemotherapy. The median Overall Survival was 17.1 months in the Immunotherapy combination group compared to 14.9 months in the chemotherapy group (HR=0.79; P=0.007). Progression Free Survival, Objective Response Rates and Duration of Response were all greater with OPDIVO® plus YERVOY® combination, compared to chemotherapy. In the Part 1b cohort with PD-L1 expression of less than 1%, Overall Survival benefit was again observed with the OPDIVO® plus YERVOY® combination, compared with chemotherapy. Grade 3 and 4 treatment-related Adverse Events across all patients was 33% in those treated with OPDIVO® plus YERVOY® combination, 19% with single agent OPDIVO® and 36% with chemotherapy.
It was concluded that first-line treatment of patients with advanced NSCLC with a combination of two immunotherapy drugs improves Overall Survival, compared to chemotherapy, and offers a chemotherapy-free first line treatment option for a subset of NSCLC patients, leaving chemotherapy for later lines of therapy. Nivolumab + low-dose ipilimumab versus platinum-doublet chemotherapy as first-line treatment for advanced non–small cell lung cancer: CheckMate-227 part 1 final analysis. Peters S, Ramalingam SS, Paz-Ares L, et al. Presented at 2019 ESMO Congress; September 27 to October 1, 2019; Barcelona, Spain. Abstract LBA4.
Updated Analysis: KEYTRUDA® Doubles Overall Survival Compared with Chemotherapy in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019, about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced Non Small Cell Lung Cancer (NSCLC) have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.
KEYNOTE-024 is an open-label, randomized phase III trial in which KEYTRUDA® administered at a fixed dose was compared with investigator’s choice of cytotoxic chemotherapy, as first line therapy, for patients with advanced NSCLC, with tumor PD-L1 expression of 50% or greater. Three hundred and five (N=305) treatment naïve patients with advanced NSCLC and PD-L1 expression on at least 50% of tumor cells, were randomly assigned in a 1:1 ratio to receive either KEYTRUDA® (N=154) or chemotherapy (N=151). Enrolled patients had no sensitizing EGFR mutations or ALK translocations. Treatment consisted of KEYTRUDA® administered at a fixed dose of 200 mg IV every 3 weeks for up to 2 years or the investigator’s choice of platinum-based chemotherapy for 4-6 cycles. Pemetrexed (ALIMTA®) based therapy was permitted only for patients who had non-squamous tumors and these patients could receive ALIMTA® maintenance therapy after the completion of combination chemotherapy. Patients in the chemotherapy group who experienced disease progression were allowed to cross over to the KEYTRUDA® group. The Primary end point was Progression Free Survival (PFS) and Secondary end points included Overall Survival (OS), Objective Response Rate (ORR) and Safety.
It was previously reported that at a median follow up of 11.2 months, the median PFS was 10.3 months in the KEYTRUDA® group versus 6 months in the chemotherapy group (HR=0.50; P<0.001). However, median OS had not been reached in the KEYTRUDA® group at the time of that analysis. The Independent Data and Safety Monitoring Committee (IDMC) based on these results recommended stopping the trial early, to allow for use of KEYTRUDA® in patients randomly assigned to chemotherapy. Eighty two patients (N=82) assigned to chemotherapy, met criteria to cross over to the KEYTRUDA® group, upon progression.
This publication is an updated analysis of the KEYNOTE-024 study, after a median follow-up of 25.2 months. The median OS was 30 months in the KEYTRUDA® group and 14.2 months in the chemotherapy group (HR=0.63; P=0.002). When adjusted for crossover, the OS benefit was maintained and the Hazard Ratio for OS among KEYTRUDA® group versus chemotherapy group was 0.49. Further, more patients in the KEYTRUDA® group achieved 12-month OS (70.3% versus 54.8%), and an ORR response (45.5% versus 29.8%), compared to the chemotherapy group respectively. The ORR among those who crossed over to KEYTRUDA®, was 20.7%. The median Duration of Response has not yet been reached for patients assigned to KEYTRUDA® and also for those who crossed over to KEYTRUDA®. For those assigned chemotherapy, the median Duration of Response was 7.1 months. Patients in the KEYTRUDA® group had lower rates of Grade 3-5 adverse events, compared to those in the chemotherapy group (31.2% versus 53.3%), as well as a lower rate of any-grade adverse events (76.6% versus 90%).
It was concluded that in this updated analysis of KEYNOTE-024, KEYTRUDA® continued to provide improved Overall Survival benefit, inspite of the high rate of crossover, with lower rates of Adverse Events, when compared to chemotherapy, among patients with metastatic NSCLC and high PD-L1 expression. The authors added that these updated long term results support KEYTRUDA® monotherapy as a standard-of-care regimen for first line treatment of advanced NSCLC with PD-L1 expression of 50% or greater and without EGFR/ALK alterations. Updated Analysis of KEYNOTE-024: Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score of 50% or Greater. Reck M , Rodríguez–Abreu D, Robinson AG, et al. J Clin Oncol 2019;37:537-546
SBRT Superior to Standard Radiotherapy in Stage I Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019, about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. Approximately 15% of patients present with early stage (T1-2 N0) disease, and these numbers are likely to increase with the implementation of Lung Cancer screening programs. Patients with early stage disease unless medically unfit, undergo surgical resection with a curative intent. Those who are not surgical candidates are often treated with conventional Radiation Therapy, over a period of 4 to 6 weeks.
Stereotactic Body Radiation Therapy (SBRT) is a non-surgical procedure that allows delivery of significantly higher doses of precisely focused radiation to the tumor, compared to conventional Radiation Therapy, with less collateral damage to the surrounding normal tissue. The technologies used for SBRT include GAMMA KNIFE® which uses highly focused gamma rays, Proton Beam therapy which uses ionized Hydrogen or Protons, Linear Accelerator (LINAC) and CYBER KNIFE® which use Photons, to target the tumor tissue. Because SBRT is fractionated and delivered over 1-5 days, the short-and long-term side effects of radiation therapy are decreased and may allow higher total dosage to be given. Previously published single-arm trials have shown high local control with SBRT, with no significant difference in Overall Survival, compared with conventional Radiotherapy. This Phase III trial was conducted to prospectively assess the effect of SBRT on local control, Overall Survival, toxicity and Quality of Life.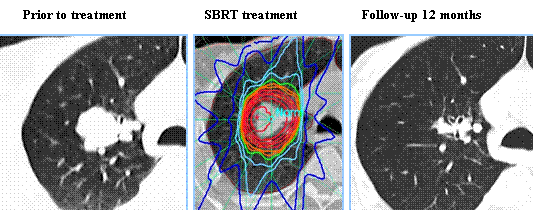
In this multicentre, randomized, Phase III trial, 101 eligible patients with biopsy proven Stage I (T1-T2aN0M0) NSCLC, diagnosed on the basis of FDG PET, who were medically inoperable or had refused surgery, were enrolled. Patients were randomly assigned in a 2:1 ratio to SBRT (54 Gy in three 18 Gy fractions, or 48 Gy in four 12 Gy fractions if the tumor was less than 2 cm from the chest wall)-(N=66) or standard Radiotherapy (66 Gy in 33 daily 2 Gy fractions or 50 Gy in 20 daily 2.5 Gy fractions (N=35), based on institutional preference. The tumor had to be non-central and peripherally located, at least 1 cm in the mediastinum and 2 cm from the bifurcation of the lobar bronchi. Patients were stratified by T stage and operability (medically operable but refused surgery versus inoperable). The Primary endpoint was time to local treatment failure and Secondary endpoints included Overall Survival, treatment related toxicity and Quality of Life. The median follow up for local treatment failure was 2.1 years for standard Radiotherapy group and 2.6 years for those patients assigned to SBRT.
Local treatment failure was noted in 14% of patients in the SBRT group whereas 31% of patients in the standard Radiotherapy group progressed locally. Freedom from local treatment failure was significantly improved the SBRT group compared with a standard radiotherapy group (HR=0.32, P=0.0077). Median time to local treatment failure was not reached in either group. Median Overall Survival was 5 years in the SBRT group and 3 years in the standard Radiotherapy group (HR=0.53; P=0.027). Overall Survival at 2 years was 77% for those receiving SBRT and 59% for those in the standard Radiotherapy group. Treatment related toxicities were low in both groups and there were no significant differences in Quality of Life between the treatment groups.
It was concluded that in patients with inoperable peripherally located Stage 1 NSCLC, compared with standard Radiotherapy, SBRT resulted in superior local control of the primary disease without an increase in major toxicity, and improvement in Overall Survival. The authors added that these findings suggest that SBRT should be the treatment of choice for this patient group. Stereotactic ablative radiotherapy versus standard radiotherapy in stage 1 non-small-cell lung cancer (TROG 09.02 CHISEL): a phase 3, open-label, randomised controlled trial. Ball D, Tao Mai G, Vinod S, et al. Lancet Oncol 2019;20:494-503
Survival Benefit with KEYTRUDA® After Locally Ablative Therapy (LAT) for Oligometastatic NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019, about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas.
It is estimated that approximately 7% of patients with NSCLC present with a limited number of metastatic foci (oligometastatic). Several retrospective studies have shown that the use of Locally Ablative Therapy (LAT) to all sites of disease in oligometastatic NSCLC is associated with a significant improvement in Progression Free Survival (PFS) and Overall Survival (OS), when compared with historical data. Preclinical evidence had suggested that chemotherapy and radiotherapy may upregulate PD-L1 expression in tumor cells. Therefore, incorporating immunotherapy along with LAT has been an area of active research. In the PACIFIC trial, consolidation therapy with PD-L1 inhibitor IMFINZI® (Durvalumab), following chemoradiation, significantly improved PFS and OS among patients with locally advanced NSCLC suggesting that there is a strong biological rationale for the use of immunotherapy in patients with minimal residual disease state.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. The primary objective of this study was to evaluate whether the addition of KEYTRUDA® after Locally Ablative Therapy (LAT) improves outcomes among patients with oligometastatic NSCLC, compared with historical data.
The authors conducted a single arm Phase II trial at an academic referral cancer center, and 51 eligible patients with oligometastatic NSCLC (no more than 4 metastatic sites) were enrolled. Enrolled patients had oligometastatic disease at diagnosis (synchronous disease) or who developed oligometastatic disease after initial definitive therapy (metachronous disease). There was no limit on the number of prior therapies, although patients could not have received prior therapy with a Programmed Death 1 (PD-L1) inhibitor. Any form of Locally Ablative Therapy (LAT) was acceptable and LATs included Surgery, Chemoradiotherapy, Stereotactic radiotherapy, and/or Interventional ablation. Forty five of the 51 patients enrolled received KEYTRUDA® within 4 to 12 weeks of completing LAT. Patients received KEYTRUDA® 200 mg IV every 21 days, for 8 cycles and were allowed to continue therapy for a total of 16 cycles in the absence of progressive disease or untoward toxicities. The median age was 64 years and patients were eligible regardless of their PD-L1 or molecular target status. Thirty-two patients had adequate tissue for assessment of PD-L1 status, and 29 patients had adequate tissue for assessment of CD8 T-cell infiltration. In patients undergoing testing, 34% had results positive for PD-L1 (1% or more) and 52% had CD8 T-cell infiltration of greater than 2.5%. Patients received a median of 11 cycles of KEYTRUDA®.
The two Primary efficacy end points were Progression Free Survival (PFS) from the start of Locally Ablative Therapy (PFS-L) and PFS from the start of KEYTRUDA® therapy (PFS-P). This study was powered for comparison with historical data on the first efficacy end point. Secondary outcomes included Overall Survival, Safety, and Quality of Life, as measured by the Functional Assessment of Cancer Therapy–Lung (FACT-L) instrument.
After a median follow-up of 23.2 months for surviving patients, the median PFS from the start of Locally Ablative Therapy (PFS-L) was 19.1 months, which was a statistically significant improvement from the historical median of 6.6 months (P=0.005). The median PFS from the start of KEYTRUDA® therapy (PFS-P) was 18.7 months. The mean Overall Survival rate at 12 months was 90.9% and at 24 months was 77.5%. The Progression Free Survival from the start of Locally Ablative Therapy (PFS-L) was not influenced by PD-L1 expression or CD8 T-cell tumor infiltration. Quality of Life as measured by the FACT-L scores at cycles 8 and 16 were not significantly different from FACT-L scores at baseline.
The authors concluded that KEYTRUDA® after Locally Ablative Therapy for oligometastatic NSCLC was associated with a clinically and statistically significant improvement in Progression Free Survival, compared with historical data, without a decrement in Quality of Life. They added that the Overall Survival data is encouraging but will require further follow up. Pembrolizumab After Completion of Locally Ablative Therapy for Oligometastatic Non–Small Cell Lung Cancer: A Phase 2 Trial. Bauml JM, Mick R, Ciunci C, et al. JAMA Oncol. Published online July 11, 2019. doi:10.1001/jamaoncol.2019.1449
Low Provider Knowledge Associated with Less Lung Cancer Screening
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019, about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. In the National Lung Screening Trial (NLST) with Low Dose CT (LDCT) screening for lung cancer, there was a 20% reduction in mortality. Following the publication of the results of NLST and NCCN issued guideline in 2011, the United States Preventive Services Task Force (USPSTF) recommended Lung Cancer screening with Low Dose CT scan in high risk patients. CMS in 2015 determined that there was sufficient evidence to reimburse for this preventive service.
Despite the evidence and recommendation along with supportive public policies, screening with LDCT has not been adequately implemented in the US healthcare system. (A low-dose CT scan will typically deliver 1-4 millisieverts of radiation exposure, whereas a conventional CT scan typically delivers between 5-20 millisieverts).Health Care Providers play a key role because LDCT is currently the only cancer screening modality required by CMS to have a shared decision-making conversation with the patient for reimbursement, making it crucial for providers to have knowledge of the screening recommendations, CMS coverage criteria, and evidence for screening. Low Health Care Provider knowledge of the Lung Cancer Screening (LCS) guidelines represents a potential barrier to implementation.
The authors in this study tested the hypothesis that low provider knowledge of Lung Cancer Screening guidelines is associated with less provider-reported screening with LDCT. The researchers from February thru May 2017 surveyed/invited 625 providers who routinely perform health screenings or care for patients at high risk of lung cancer, out of whom responses from 378 providers were analyzed. Health Care Providers were emailed internet-based questionnaire and participating providers included those who practiced general Internal Medicine/Family Medicine, Pulmonology, Hematology/Oncology, and Gynecology, within an academic medical center (Vanderbilt University Medical Center [VUMC]) and its affiliated Veterans Health Administration (VHA), including hospital-based and community-based practices. A medicine Grand Rounds at VUMC focused on Lung Cancer Screening (LCS) prior to this survey. Eligible providers included Attending physicians, Physicians in training, Physician Assistants, and Nurse Practitioners, who reported providing healthcare services to patients aged more than 50 in the year before the study. The questionnaire was terminated if respondents did not meet these criteria. Approximately 47% were Attending physicians, 43% were Physicians in training and 10% were Nurse Practitioners/Physician Assistants. Questionnaire Content included six multiple-choice items based on the USPSTF and CMS coverage criteria for Lung Cancer Screening. They included the following:
1) Initial age of screening eligibility (correct answer: 55 years)
2) Upper age limit at which a patient is no longer eligible for screening (correct answer: either 77 or 80 years)
3) Minimum smoking exposure (correct answer: 30 pack-years)
4) Smoking status (correct answer: current and former smokers)
5) Screening frequency (correct answer: yearly)
6) Screening for patients who were not surgical candidates (correct answer: no)
High LCS knowledge was defined as correctly identifying 3 major criteria to identify screening candidates: initial age of screening eligibility, minimum smoking exposure, and smoking status. Low LCS knowledge was defined as not correctly identifying these 3 criteria. The Primary outcome was self-reported order/referral of LDCT within the past year. Secondary outcomes were self-reported ordering of non-recommended LCS tests such as chest X Ray and sputum cytology.
On analysis it was noted that 59% of the providers reported ordering/referring for LDCT within the past year. Overall 62% of the provider’s demonstrated low LCS knowledge and the odds of ordering/referring for LDCT were 76% lower for providers with low LCS knowledge than for those with high LCS knowledge. Providers with low LCS knowledge had a 2.7 higher odds of ordering a screening chest radiograph, than providers with high LCS knowledge. The ordering/referring rates for LDCT were highest among general Internal Medicine/Primary Care Providers (75.9%), followed by Pulmonologists (74.4%), Hematologists/Oncologists (39.7%), and Gynecologists (2.1%). After adjusting for other variables, all provider types were less likely than general Internists/Primary Care Providers to order/refer for LDCT.
It was concluded that results of this study suggests that the referring provider knowledge of LCS guidelines is low and providers with low guideline knowledge were less likely to order LDCT lung screening. The authors recommended that strategies to advance effective evidence-based LCS should incorporate provider education and healthcare system interventions, to facilitate the appropriate identification of candidates for Lung Cancer Screening. Low Provider Knowledge Is Associated With Less Evidence-Based Lung Cancer Screening. Lewis JA, Chen H, Weaver KE, et al. J Natl Compr Canc Netw 2019;17:339-346
Late Breaking Abstract - ASCO 2019: Five-Year Survival Data for KEYTRUDA® in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019 about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression, and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.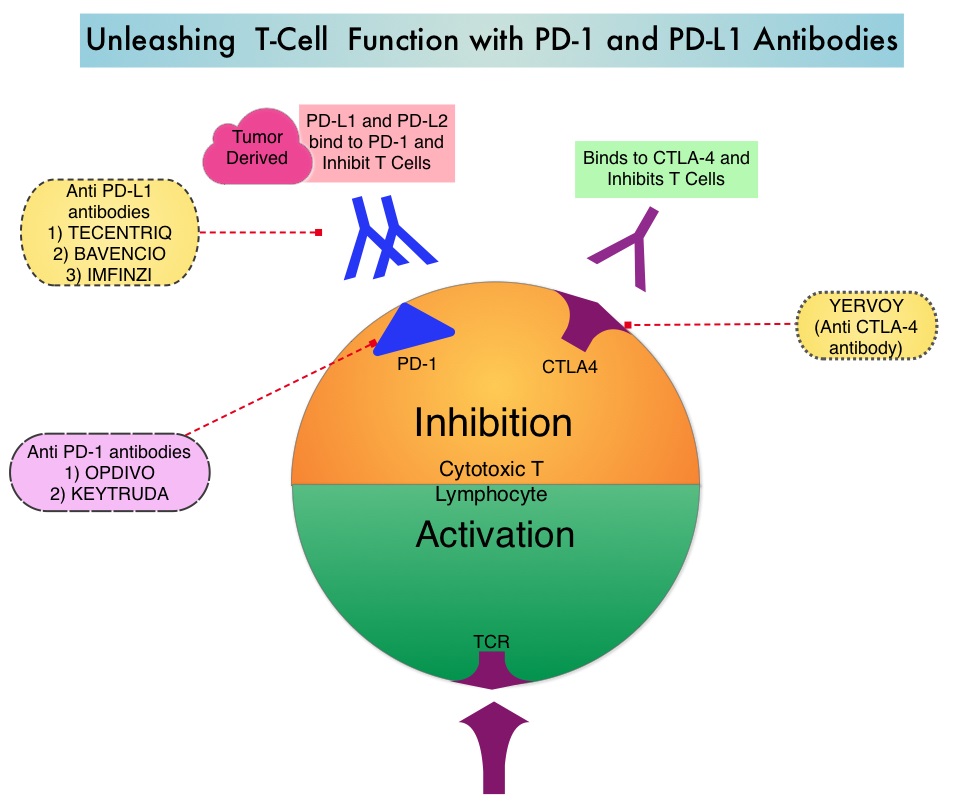
The FDA approved KEYTRUDA® for the first-line treatment of patients with Stage III Non-Small Cell Lung Cancer (NSCLC) who are not candidates for surgical resection or definitive chemoradiation, as well as those with metastatic NSCLC whose tumors express PD-L1 (Tumor Proportion Score-TPS of 1% or more), as determined by an FDA-approved test. KEYTRUDA® is also approved for the first-line treatment of advanced NSCLC with high PD-L1 expression (Tumor Proportion Score of 50% or more), based on KEYNOTE-024 trial, as well as in combination with Pemetrexed and Carboplatin, as first-line treatment of patients with metastatic non-squamous NSCLC, based on KEYNOTE-021 study. It is also indicated for previously treated advanced NSCLC with a much lower level of PD-L1 expression such as PD-L1 Tumor Proportion Score of 1% or higher, based on KEYNOTE-010 trial.
The authors in this publication presented the 5-year Overall Survival (OS) for patients enrolled in the Phase 1b KEYNOTE-001 study, which was the first trial evaluating KEYTRUDA® in advanced NSCLC. In this trial, 550 patients were enrolled of whom 101 patients were treatment-naïve (N=101) and 449 patients were previously treated (N=449). Patients received KEYTRUDA® 2 mg/kg IV every 3 weeks or KEYTRUDA® 10 mg/kg IV every 2 or 3 weeks. The protocol in the recent years was changed to a straight dose of KEYTRUDA® 200 mg IV every 3 weeks, which is the typical regimen used in clinical practice. The Primary endpoint was Objective Response Rate (ORR). Secondary endpoints included Progression Free Survival (PFS), Overall Survival (OS) and Duration of Response (DOR). The median follow up was 60.6 months and 18% of participants (N=100) were still alive at that point.
The 5-year OS in the treatment-naïve patients (N=101) was 23.2% and 15.5% in previously treated patients (N=449). In treatment-naive patients, the 5-year OS rate among patients whose tumors expressed PD-L1 expression of 50% or more was 29.6%, compared with 15.7% with PD-L1 expression levels below 50%. In patients who had received previous treatment, the 5-year OS rate among patients whose tumors expressed PD-L1 expression of 50% or more was 25% compared with 12.6% with PD-L1 expression levels between 1% and 49%. Only 3.5% of people with PD-L1 expression levels below 1% were alive after 5 years. The investigator-reported ORR was 41.6% in treatment-naïve patients and 22.9% in previously treated patients. Median Duration of Response was 16.8 months and 38.9 months respectively. Immune-mediated adverse events were reported in 17% of patients at 5 years. Hypothyroidism was the most commonly reported immune-mediated adverse event, followed by pneumonitis, hyperthyroidism and skin toxicities.
It was concluded that the 5-year data from the KEYNOTE-001 trial showed that treatment with KEYTRUDA® was safe and effective and substantially increased Overall Survival in patients with advanced NSCLC. These data provide the longest efficacy and safety follow-up for NSCLC patients treated with KEYTRUDA®. Five-year long-term overall survival for patients with advanced NSCLC treated with pembrolizumab: Results from KEYNOTE-001. Garon EB, Hellmann MD, Costa EC, et al. J Clin Oncol. 2019;37(suppl; abstract LBA9015).
FDA Lowers PD-L1 Expression Threshold for KEYTRUDA® and Expands Indication for Frontline Treatment of NSCLC
SUMMARY: The FDA on April 11, 2019, approved KEYTRUDA® (Pembrolizumab) for the first-line treatment of patients with Stage III Non-Small Cell Lung Cancer (NSCLC) who are not candidates for surgical resection or definitive chemoradiation, as well as those with metastatic NSCLC. Patients’ tumors must have no EGFR or ALK genomic aberrations and express PD-L1 (Tumor Proportion Score-TPS of 1% or more), as determined by an FDA-approved test. Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019 about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression, and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®. The FDA approved KEYTRUDA® for the first-line treatment of advanced NSCLC with high PD-L1 expression (Tumor Proportion Score of 50% or more), based on KEYNOTE-024 trial, as well as in combination with Pemetrexed and Carboplatin, as first-line treatment of patients with metastatic non-squamous NSCLC, based on KEYNOTE-021 study. It is also indicated for previously treated advanced NSCLC with a much lower level of PD-L1 expression such as PD-L1 Tumor Proportion Score of 1% or higher, based on KEYNOTE-010 trial.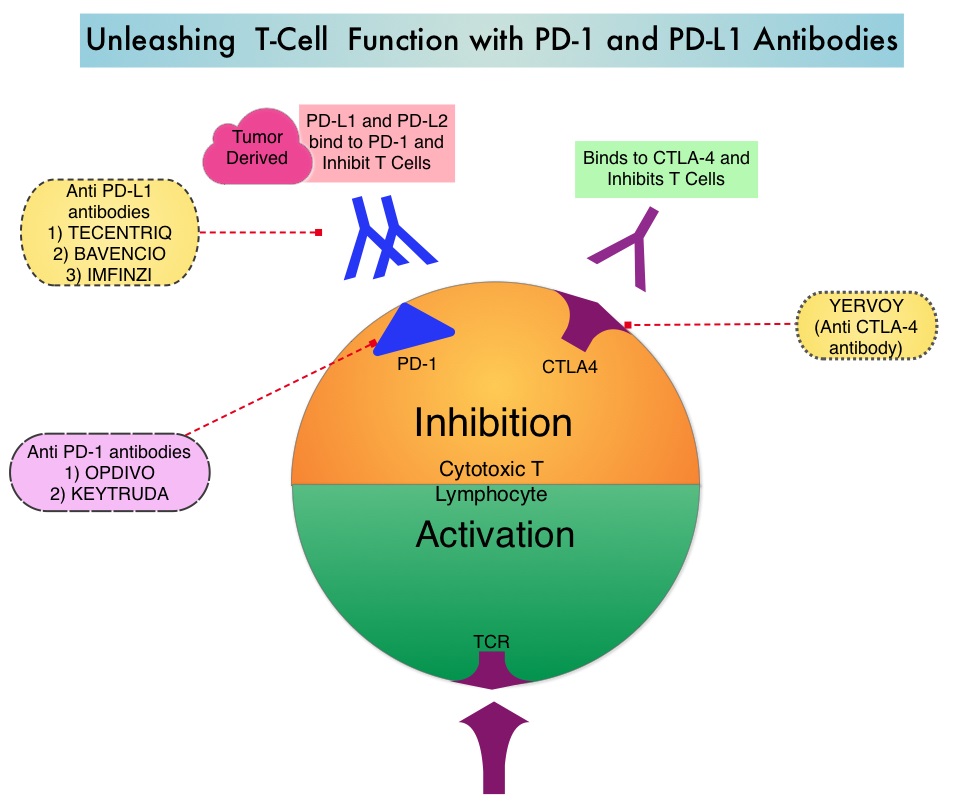
KEYNOTE-042 is a large, international, multicenter, randomized phase III trial in which 1274 patients with untreated locally advanced or metastatic NSCLC were randomly assigned to KEYTRUDA® or chemotherapy with Paclitaxel plus Carboplatin or Pemetrexed plus Carboplatin. In this study, both squamous and non-squamous cancers with PD-L1 Tumor Proportion Score (TPS) of 1% or more were included, but tumors with sensitizing Epidermal Growth Factor Receptor (EGFR) or Anaplastic Lymphoma Kinase (ALK) mutations cancers with genetic changes, that could be treated with targeted therapies such as EGFR and ALK inhibitors, were excluded. Eligible patients were randomly assigned in a 1:1 to receive either KEYTRUDA® 200 mg IV every 3 weeks for up to 35 cycles or investigator’s choice of up to 6 cycles of chemotherapy with Paclitaxel plus Carboplatin or Pemetrexed plus Carboplatin, with optional Pemetrexed maintenance for non-squamous NSCLC. Patients were divided into 3 treatment groups based on their PD-L1 Tumor Proportion Score (TPS): TPS 50% or more (N=599), TPS 20% or more (N=818), and TPS 1% or more (N=637). Each PD-L1 expression group had equal numbers of patients receiving KEYTRUDA® and chemotherapy. The Primary end points were Overall Survival (OS) in patients with TPS 50% or more, 20% or more, and 1% or more.
At a median follow up of 12.8 months, 13.7% of patients were still receiving KEYTRUDA® compared with 4.9% on Pemetrexed maintenance therapy. It was noted that KEYTRUDA® was significantly superior to chemotherapy in all PD-L1 expression subsets. In patients with a PD-L1 TPS 50% or more, the median OS with KEYTRUDA® was 20 months versus 12.2 months for chemotherapy (HR=0.69, P=0.0003), for patients with PD-L1 TPS 20% or more, the median OS was 17.7 months versus 13 months respectively (HR=0.77, P=0.002), and for those with PD-L1 TPS 1% or more, the median OS was 16.7 months versus 12.1 months respectively (HR=0.81, P = 0.0018). The Response Rates (RR) were also higher among patients who received KEYTRUDA®, with RR of 39.5% for KEYTRUDA® versus 32% for chemotherapy in patients with a TPS 50% or more, 33.4% and 28.9% respectively in patients with TPS 20% or more and 27.3% and 26.5%, respectively, among patients with TPS of 1% or more. The duration of response was also superior with KEYTRUDA® in all three PD-L1 subgroups compared to chemotherapy (20.2 months versus 8-11 months). Patients receiving KEYTRUDA® experienced fewer severe Adverse Events, compared with chemotherapy (17.8% versus 41%).
The authors concluded that this is the largest clinical trial of KEYTRUDA® as a stand-alone therapy, and is the first study with a Primary end point of OS to demonstrate superiority of KEYTRUDA® over platinum-based chemotherapy, in patients with previously untreated locally advanced/metastatic NSCLC, without sensitizing EGFR or ALK alterations and a PD-L1 TPS of 1% or more. These data confirmed the benefit of KEYTRUDA® monotherapy as a standard first-line treatment, for PD-L1-expressing locally advanced Stage III as well as metastatic NSCLC. KEYTRUDA® monotherapy is now a new treatment option for more patients with NSCLC, including those for whom combination therapy may not be appropriate. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Mok TS, Wu Y-L, Kudaba I, et al. The Lancet. Published: April 04, 2019. DOI: https://doi.org/10.1016/S0140-6736(18)32409-7
Liquid Biopsy Accurate, Reliable and Rapid in Identifying Biomarker Mutations in Newly Diagnosed Advanced Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2019 about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Patients with newly diagnosed metastatic NSCLC are often tested for guideline-recommended genomic biomarkers which include both predictive biomarker mutations such as EGFR, ALK, ROS1, BRAF, RET, MET, ERBB2, as well as prognostic biomarker mutation such as KRAS.
The application of precision medicine with targeted therapy requires detection of molecular abnormalities in a tissue biopsy specimen. However, if testing is not done with a comprehensive assay, such as Next-Generation Sequencing and is done in successive steps one test after another, tissue sample can be depleted, with not enough tissue left for testing of all biomarkers. Following progression or recurrence, archived biopsy specimens may not be helpful, as it is important to identify additional mutations in the tumor at the time of recurrence or progression, in order to plan appropriate therapy. Further, recurrent tumors may be inaccessible for a safe biopsy procedure or the clinical condition of the patient may not permit a repeat biopsy. Additionally, the biopsy itself may be subject to sampling error due to tumor heterogeneity. Genotyping circulating cell-free tumor DNA (cfDNA) in the plasma can potentially overcome the shortcomings of repeat biopsies and tissue genotyping, allowing the detection of many more targetable gene mutations, thus resulting in better evaluation of the tumor genome landscape.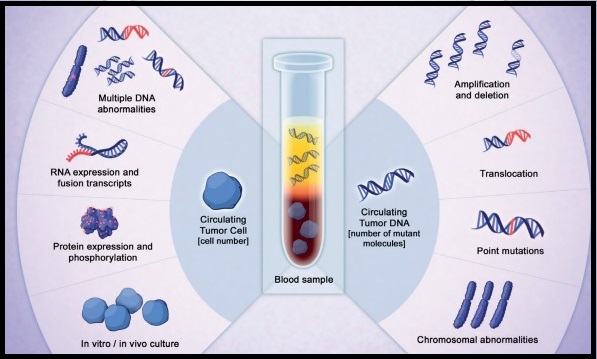
The Noninvasive versus Invasive Lung Evaluation (NILE) trial is a prospective, multicenter study conducted to demonstrate the noninferiority of comprehensive cell-free DNA (cfDNA) relative to standard-of-care traditional tissue genotyping tests, to identify guideline-recommended genomic biomarkers, in patients with metastatic NSCLC. The authors in this study enrolled 282 newly diagnosed patients at 28 North American centers, with previously untreated, nonsquamous, metastatic NSCLC undergoing standard-of-care tissue genotyping. Enrolled patients submitted a pretreatment blood sample for cfDNA analysis utilizing a CLIA-certified comprehensive 73-gene next generation sequencing panel (Guardant360®). Over 80% of the enrolled patients were white and over 50% were female.
The liquid biopsy utilizing Guardant360®, detected biomarker mutations at a rate similar to standard-of-care tissue genotyping tests, in the enrolled patients, meeting the Primary study objective. At least one of the guideline-recommended genomic biomarkers was detected in 60 patients (21.3%) using tissue-based tests and in 77 patients (27.3%) by cfDNA utilizing Guardant360® (P<0.0001). The detection rate was increased by 48% when Guardant360® was utilized for cfDNA analysis and this included those with negative, not assessed, or Quantity Not Sufficient (QNS) results in tissue. In addition, the Positive Predictive Value was 100% for cfDNA versus tissue genotyping, for FDA approved targets such as EGFR, ALK, ROS1, and BRAF mutations. There are agents already approved by the FDA to treat this patient population. The median turnaround time was significantly lower for cfDNA, compared to tissue genotyping (9 versus 15 days; P <0.0001).
The authors concluded that in this largest cfDNA study among patients with previously untreated advanced NSCLC, cfDNA successfully detected seven biomarker mutations noninvasively, significantly faster than tissue genotype testing, and was also able to rescue biomarker mutation positive patients who had non-diagnostic tissue results. They added that the findings in this study confirms similar findings from Europe and demonstrates the clinical utility of cfDNA in newly diagnosed metastatic NSCLC. Clinical utility of comprehensive cell-free DNA (cfDNA) analysis to identify genomic biomarkers in newly diagnosed metastatic non-small cell lung cancer (mNSCLC). Leighl N, Page RD, Raymond VM, et al. Presented at: AACR Annual Meeting April 2, 2019; Philadelphia, USA.
Baseline Corticosteroid Use at Start of PD-1/PD-L1 Inhibitor Therapy Negatively Affects Outcomes in NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Immunotherapy with PD-1 (Programmed cell Death 1) and PD-L1 (Programmed cell Death Ligand 1) inhibitors have demonstrated a clear survival benefit both as a single agent or in combination, compared with standard chemotherapy, in both treatment-naive and previously treated patients for advanced Non Small Cell Lung Cancer (NSCLC). It is now standard therapy for patients with lung cancer. Immuno-Oncology therapies unleash the T cells by blocking the Immune checkpoint proteins, thereby resulting in T cell proliferation, activation and a therapeutic response.
Patients with NSCLC often are treated with corticosteroids for a variety of reasons including fatigue, dyspnea, decreased appetite, and symptomatic brain metastases. Corticosteroids by virtue of their immunosuppressive properties can potentially effect on T-cell function and for this reason, use of these agents before the start of therapy with PD-(L)1 blockade has been a uniform exclusion criterion in clinical trials of Immune Checkpoint Blockade therapies. It is however becoming increasing clear that corticosteroids use for the management of immune-related adverse events do not seem to negatively impact outcomes. Nonetheless, there are presently no data regarding the impact of corticosteroid use at baseline, on the efficacy of Immune Checkpoint Inhibitors. In this publication, the authors evaluated the potential impact of systemic corticosteroids at the start of Immune Checkpoint Blockade, on the efficacy of PD-(L)1 inhibitors.
The authors in this study identified patients with advanced NSCLC who were treated with single-agent PD-(L)1 inhibitor (Pembrolizumab, Nivolumab, Atezolizumab, or Durvalumab) from two institutions - Memorial Sloan Kettering Cancer Center (N=455) and Gustave Roussy Cancer Center (N=185), between April 2011 to September 2017. Clinical and pharmacy records were reviewed to identify corticosteroid use at the time of beginning anti-PD-(L)1 therapy. Information on the use of corticosteroids within 30 days of the start of PD-(L)1 blockade, type of corticosteroid used, indication and route of administration were collected for the MSKCC cohort. Patient characteristics, including age, gender, histology, ECOG Performance Status, and smoking history were collected for all patients. Efficacy outcomes following treatment with PD-(L)1 inhibitors blockade was determined by local radiologists and all patients were followed up until death or data lock.
It was noted that 14% (N=90) of the 640 patients treated with single-agent PD-(L)1 inhibitor received 10 mg or more of prednisone daily at the start of the treatment with a PD-(L)1 inhibitor. The most common indications for treatment with corticosteroids were dyspnea or other respiratory symptoms (33%), fatigue (21%), and brain metastases (19%). Patient characteristics were well balanced between those who did or did not receive corticosteroids, with two exceptions - patients with poor performance status and history of brain metastases were more common in those who received corticosteroids.
In the pooled cohort of patients from both participating institutions, patients receiving baseline corticosteroids compared with patients not receiving corticosteroids experienced a lower Objective Response Rate (7% versus 18%) and worse Progression Free Survival and Overall Survival (P<0.001). The authors performed a multivariable analysis in the pooled cohort (N = 640), incorporating smoking history, performance status, history of brain metastases, and corticosteroid use (Prednisone 10 mg or more versus less than 10 mg), at the start of PD-(L)1 blockade. Prednisone use 10 mg or more was associated with worse Progression Free Survival (P=0.03) and Overall Survival (P<0.001). In the Memorial Sloan Kettering Center cohort of patients, (data unavailable for the Gustave Roussy Cancer Center cohort), patients who discontinued corticosteroids 1-30 days before starting PD-(L)1 blockade had intermediate Progression Free Survival and Overall Survival compared to those who received corticosteroids on the day of PD-(L)1 blockade initiation and those who received no corticosteroids within 30 days of the start of therapy.
The authors concluded that among patients with Non Small Cell Lung Cancer treated with PD-(L)1 blockade, baseline corticosteroid use of 10 mg or more of prednisone equivalent was associated with inferior outcomes. Clinicians should exercise caution and minimize the use, duration, and dose of corticosteroids if immunotherapy with PD-(L)1 blockade is a future consideration. Impact of Baseline Steroids on Efficacy of Programmed Cell Death-1 and Programmed Death-Ligand 1 Blockade in Patients With Non-Small-Cell Lung Cancer. Arbour KC, Mezquita L, Long N, et al. J Clin Oncol. 2018;36:2872-2878
The International Association for the Study of Lung Cancer Issues Statement on Lung Cancer Screening: CALL TO ACTION
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer related mortality in the United States. Lung cancer is a growing global epidemic with 1.6 million deaths annually. Over 60% of individuals present with advanced disease at the time of diagnosis and this can result in poor outcomes. Early detection can however lead to lowered mortality. Implementing a validated tool to reliably detect early stage, curable lung cancer has been a priority of the International Association for the Study of Lung Cancer (IASLC), in its mission to conquer thoracic cancers worldwide.
The IASLC on October 25, 2018 issued a statement on lung cancer screening with Low-Dose Computed Tomography (LDCT), based on results from the Dutch-Belgian NELSON lung cancer screening trial presented at the IASLC 19th World Conference on Lung Cancer (WCLC) in Toronto, Canada. The IASLC is the only global organization dedicated solely to the study of lung cancer and other thoracic malignancies and includes more than 7,500 lung cancer specialists across all disciplines in over 100 countries.
EVIDENCE:
The National Lung Cancer Screening Trial (NLST) demonstrated that annual lung cancer screening with Low-Dose CT (LDCT) reduced lung cancer mortality by 20% and overall mortality by 7% compared with controls. Based on the NLST results, NCCN issued guidelines recommending LDCT in 2011, USPSTF (United States Preventive Services Task Force) recommended lung cancer screening with LDCT in high risk patients in 2013 and Low-Dose CT screening was approved in the United States for those at high risk (between the ages of 55 and 77 and a smoking history of 30 pack-years or more and not have quit within the past 15 years).
The Dutch-Belgian Lung Cancer Screening Trial (NELSON) is Europe’s largest lung cancer screening trial and enrolled 15,792 individuals at high risk for lung cancer. Data from this study was presented at the World Conference on Lung Cancer this year which decisively confirmed that annual lung cancer screening with Low-Dose CT in high-risk patients ((age 50-74 years, more than 10 cigarettes/day for more than 30 years or more than 15 cigarettes/day for more than 25 years), reduced lung cancer deaths by 26% in men and up to 61% in women.
RECOMMENDATIONS:
With two trials from the United States and Europe demonstrating significant mortality reduction in high risk, tobacco-exposed populations, IASLC emphasizes that early detection must be routinely provided along with best-practice smoking cessation, to enable optimal health outcomes in the setting of individuals who continue to consume tobacco products. Acknowledging that for implementation of Low-Dose CT screening worldwide, each national health service has the authority to decide its own course of action, IASLC has urged its members and others around the world to implement screening programs that incorporate a multidisciplinary group of experts and use best practice in screening care, with focus on the following:
• Identification of high-risk individuals
• Acquisition of consistent high-quality images (from Low-Dose CT) and incorporation of radiologic guidelines, including definitions for positive versus negative results
• Use of defined clinical workup for indeterminate nodules and for pathology reporting of nodules
• Incorporation of a defined process for surgical or other diagnostic interventions of suspicious nodules
• Integration of smoking cessation into lung cancer CT screening programs
It was concluded that based on the data from these two large, well designed US and European randomized trials, the WCLC committee’s screening experts came to an unanimous consensus that now is the time for international leaders, governments, health care systems and other stakeholders to implement global lung cancer screening programs, as they do for breast cancer (mammography) and colon cancer (colonoscopy), which save the thousands of lives. https://www.iaslc.org/news/iaslc-issues-statement-lung-cancer-screening-low-dose-computed-tomography
IMFINZI® after Chemoradiotherapy Significantly Improves Overall Survival in Stage III NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), approximately 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas, and 10% are Large cell carcinomas.
Approximately one third of all patients with NSCLC have stage III, locally advanced disease at the time of initial presentation. Worldwide, about 500,000 patients are diagnosed with unresectable, stage III NSCLC, each year. These patients include those with locally advanced primary tumors with tumor invading the vital mediastinal organs, as well as those with involvement of locoregional mediastinal lymph nodes. These patients are often treated with platinum-based doublet chemotherapy with concurrent radiation and have a median Progression Free Survival (PFS) of approximately 8 months and 5 year survival of only 15%. There is hence a significant unmet need for this patient group, with no major treatment advances thus far.
Preclinical evidence had suggested that chemotherapy and radiotherapy may upregulate PD-L1 expression in tumor cells. IMFINZI® (Durvalumab) is a selective, high-affinity, human IgG1 monoclonal antibody, that blocks the binding of Programmed Death Ligand 1 (PD-L1) to Programmed Death 1 (PD-1) receptor and CD80, thereby unleashing the T cells to recognize and kill tumor cells. IMFINZI® showed encouraging antitumor activity in an early phase clinical study involving multiple advanced solid tumors, including stage IIIB or IV NSCLC.
PACIFIC trial is a randomized, double-blind, international, phase III study in which IMFINZI® as consolidation therapy was compared with placebo, in patients with stage III, locally advanced, unresectable NSCLC, that had not progressed following platinum-based chemoradiotherapy. Eligible patients received two or more cycles of platinum-based doublet chemotherapy concurrently with definitive radiation therapy (54-66 Gy). Following completion of concurrent chemoradiation treatment, 713 patients were randomized, of whom 709 patients in a 2:1 ratio received consolidation treatment, within 6 weeks after completion of chemoradiation, with IMFINZI® 10 mg/kg every 2 weeks (N=473) or placebo (N=236), for up to 12 months. The median age was 64 years, and the majority of patients were men (70%) and 46% had a squamous histology. The co-Primary end points were Progression Free Survival (PFS) and Overall Survival (OS). Secondary end points included 12-month and 18-month PFS rates, Objective Response Rate (ORR), Duration of Response, time to death or distant metastasis, and safety.
The authors had previously reported the results of the first preplanned interim analysis, after a median follow up of 14.5 months. The median PFS from randomization to consolidation treatment was 16.8 months with IMFINZI® versus 5.6 months with placebo (HR=0.52; P<0.001). This meant a 11.2-month improvement in PFS with IMFINZI® versus placebo, and a 48% decrease in the probability of disease progression with IMFINZI®. This improvement was consistent across all patient subgroups that were analyzed.
The authors in this publication report the results for the second Primary end point of Overall Survival. At a median follow up of 25.2 months, the 24-month Overall Survival rate was 66.3% in the IMFINZI® group and 55.6% in the placebo group, suggesting a significantly prolonged Overall Survival with IMFINZI® when compared with placebo and a 32% reduction in the risk of death (HR for death=0.68; P=0.0025). The Overall Survival benefit with IMFINZI®, was observed across all the prespecified subgroups. In this updated analysis, the PFS was similar to those previously reported, with a median duration of 17.2 months in the IMFINZI® group and 5.6 months in the placebo group (HR=0.51). The median time to death or distant metastasis was 28.3 months in the IMFINZI® group and 16.2 months in the placebo group (HR=0.53). Approximately 30% of the patients in the IMFINZI® group and 26% of those in the placebo group had grade 3 or 4 adverse events of any cause, and 15% and 10% of the patients respectively, discontinued the trial regimen because of adverse events.
The authors concluded that in this updated analysis of the PACIFIC trial, the Primary end point of Overall Survival was significantly longer with IMFINZI® than with placebo, among patients with unresectable stage III NSCLC, in all the prespecified subgroups. The updated results for Secondary end points, including the time to death or distant metastasis, the incidence of new lesions, and the Objective Response Rate, were similar to those that were previously reported. The authors commented that PACIFIC trial is the first study to demonstrate a survival advantage for unresectable Stage III NSCLC, supporting this regimen as the standard of care. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. Antonia SJ, Villegas A, Daniel D, et al. [published online ahead of print September 25, 2018]. N Eng J Med. doi: 10.1056/NEJMoa1809697.
First Line KEYTRUDA® plus Chemotherapy Improves Overall Survival in Advanced Squamous NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), approximately 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas, and 10% are Large cell carcinomas. Non Small Cell Lung Cancer patients with Squamous Cell histology have been a traditionally hard- to-treat, patient group, and less than 15% of patients with advanced Squamous NSCLC survive a year after diagnosis and less than 5% of patients survive for five years or longer.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. The FDA approved KEYTRUDA® for the first-line treatment of advanced NSCLC with high PD-L1 expression (Tumor Proportion Score of 50% or more), as well as in combination with Pemetrexed and Carboplatin, as first-line treatment of patients with metastatic nonsquamous NSCLC and for previously treated advanced NSCLC with a PD-L1 Tumor Proportion Score of 1% or more. Currently, KEYTRUDA® currently is the only FDA approved immunotherapy for initial treatment of NSCLC as monotherapy (KEYNOTE-024) or in combination with chemotherapy (KEYNOTE-189).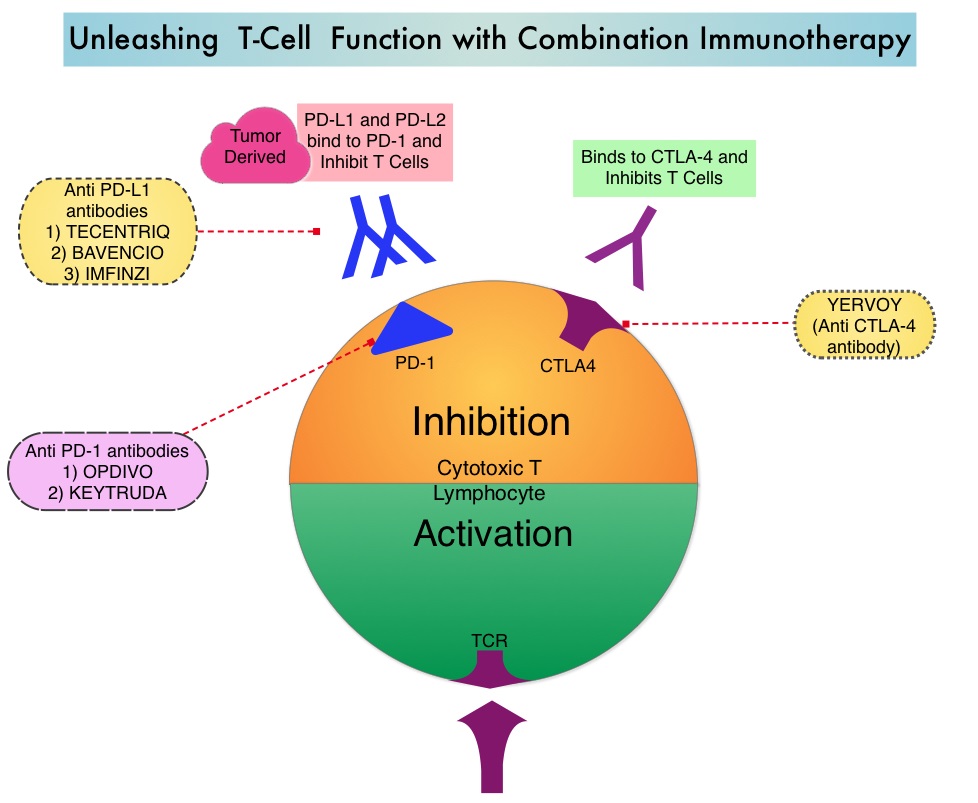
KEYNOTE-407 is a global, double-blind, placebo-controlled, phase 3 trial which compared KEYTRUDA® plus chemotherapy with placebo plus chemotherapy in patients with squamous NSCLC of any level of PD-L1 expression. In this study, 559 patients with untreated metastatic, squamous NSCLC were randomly assigned, in a 1:1 ratio to receive KEYTRUDA® 200 mg IV along with Carboplatin AUC 6 and either TAXOL® (Paclitaxel) 200 mg/m2 IV or ABRAXANE® (nab-paclitaxel) 100 mg/m2 IV days 1, 8 and 15. every 3 weeks for 4 cycles (N=278) or placebo with the same chemotherapy regimen for 4 cycles (N=281). Patients in the experimental arm following the first 4 cycles continued KEYTRUDA® every 3 weeks, for an additional 31 cycles, whereas the control group received placebo. Patients in the placebo-combination group were eligible to cross over to receive KEYTRUDA® monotherapy and 42.8% of patients assigned to the placebo plus chemotherapy group who discontinued chemotherapy went on to receive subsequent anti PD-1 or anti PD-L1 therapy and 75 patients received KEYTRUDA® monotherapy as part of in-study crossover. Patients were stratified according to the PD-L1 Tumor Proportion Score (1% or less versus more than 1%), choice of Taxane (Paclitaxel versus nab-Paclitaxel), and geographic region of enrollment. Tumor Proportion Score is the percentage of tumor cells with membranous PD-L1 staining, with less than 1% indicating PD-L1 negative score. Both treatment groups were well balanced. The co-Primary end points were Overall Survival and Progression Free Survival and the Secondary end points included Response Rate, Duration of Response and Safety. The effects of PD-L1 expression on Overall Survival (OS), Progression Free Survival (PFS), and Objective Response Rate (ORR) were prespecified exploratory end points.
At the second interim analysis, after a median follow-up of 7.8 months, the median Overall Survival was 15.9 months in the KEYTRUDA® combination group and 11.3 months in the placebo combination group (HR=0.64; P<0.001). This meant that there was a 36% reduction in the risk of death with the addition of KEYTRUDA® to chemotherapy. This OS benefit was consistently seen regardless of the level of PD-L1 expression. The median PFS was 6.4 months in the KEYTRUDA® combination group and 4.8 months in the placebo combination group (HR=0.56; P<0.001) and this suggested that the risk of disease progression or death was 44% lower with the addition of KEYTRUDA® to chemotherapy. The PFS benefit with the addition of KEYTRUDA® to chemotherapy was observed in all prespecified subgroups with incremental improvements noted with increasing PD-L1 Tumor Proportion Score. The Objective Response Rate was also significantly higher in the KEYTRUDA® chemotherapy group compared to the placebo chemotherapy group (59.4% versus 38%; P=0.0004), with a median time to response of 1.4 months and median Duration of Response of 7.7 months versus 4.8 months, respectively. Grade 3 or higher adverse events were similar in both treatment groups. Treatment discontinuation due to adverse events was more frequent in the KEYTRUDA® combination group (13.3% versus 6.4%).
It was concluded that inpatients with previously untreated metastatic, squamous NSCLC, the addition of KEYTRUDA® to chemotherapy resulted in significantly longer Overall Survival and Progression Free Survival than chemotherapy alone, and should become a new standard of care for squamous NSCLC. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. Paz-Ares L, Luft A, Vicente D, et al. for the KEYNOTE-407 Investigators. September 25, 2018. DOI: 10.1056/NEJMoa1810865
Hyperprogressive Disease after Immunotherapy in Advanced Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Immunotherapy with PD-1 (Programmed cell Death 1) and PD-L1 (Programmed cell Death Ligand 1) inhibitors have demonstrated a clear survival benefit both as a single agent or in combination, compared with standard chemotherapy, in both treatment-naive and previously treated patients for advanced Non Small Cell Lung Cancer (NSCLC). Immuno-Oncology therapies unleash the T cells by blocking the Immune checkpoint proteins, thereby resulting in T cell proliferation, activation and a therapeutic response.
Recent reports of an acceleration of tumor growth during immunotherapy, defined as HyperProgressive Disease (HPD), has been observed in 9% of advanced malignancies and in 29% of patients with Head and Neck cancer treated with PD-1/PD-L1 inhibitors. It has been postulated that high level of interferon gamma (IFN-gamma) usually released by PD-1 blockade may have detrimental effects on immunity. Alternatively PD-1/PD-L1 blockade may upregulate Interleukin 6, Interleukin 17, and neutrophil axis, generating a potent aberrant inflammation, responsible for immune escape and accelerated growth.
HyperProgressive Disease should be differentiated from Pseudoprogression. The later is defined as progressive disease, followed by Complete Response and/or Partial Response or Stable Disease longer than 6 months. The Tumor Growth Rate (TGR) estimates the increase in tumor volume over time based on two Computed Tomography (CT) scan measurements. TGR can be used to quantitatively assess tumor dynamics and kinetics during treatment and can be specifically applied to identify the subset of patients experiencing HPD.
This study was conducted to investigate whether HPD is observed in patients with advanced NSCLC treated with PD-1/PD-L1 inhibitors compared with single-agent chemotherapy and whether there is an association between treatment and HPD. This multicenter, retrospective study included 406 consecutive eligible patients with confirmed Stage III or IV NSCLC treated with PD-1/PD-L1 inhibitors such as OPDIVO® (Nivolumab), KEYTRUDA® (Pembrolizumab), TECENTRIQ® (Atezolizumab), or IMFINZI® (Durvalumab) as monotherapy in second or later line treatment, at eight French institutions between November 2012 and April 2017. The control cohort included equivalent data collected on 59 eligible patients with advanced NSCLC, who had failed a Platinum-based regimen and received single-agent chemotherapy (Taxanes, Pemetrexed, Vinorelbine , or Gemcitabine) in 4 French institutions from August 2011, to June 2016. The median age was 50 years, over 70% of the patients had nonsquamous histology and approximately 20% of the patients had PD-L1 positive status (1% or more by IHC) confirmed. The median followup was 12.1 months. Measurable disease (defined by Response Evaluation Criteria in Solid Tumors - RECIST version 1.1) on at least two CT scans before treatment and one CT scan during treatment, was required. HyperProgressive Disease (HPD) was defined as disease progression on the first CT scan during treatment with an absolute increase in Tumor Growth Rate exceeding 50%. The Primary end point was assessment of the HyperProgressive Disease rate in patients treated with Immunotherapy or chemotherapy.
Among those treated with PD-1/PD-L1 inhibitors, HyperProgressive Disease was noted in 13.8% of patients. HPD was significantly associated with more than two metastatic sites prior to treatment with PD-1/PD-L1 inhibitors, compared with those without HPD (62.5% versus 42.6%; P=0.006). However, baseline tumor burden and number of previous lines of therapy did not make a significant difference. Patients experiencing HPD within the first 6 weeks of beginning PD-1/PD-L1 inhibitor therapy had significantly lower median Overall Survival compared with those with progressive disease without HyperProgression at the first evaluation (3.4 months versus 6.2 months; HR=2.18; P=0.003). Pseudoprogression was observed in 4.7% patients.
Among patients treated with single-agent chemotherapy, only 5.1% were classified as having HyperProgressive Disease and the median Overall Survival was 4.5 months in those with HPD and 3.9 months in other patients with progressive disease without HyperProgression at the first evaluation (P=0.60).
The authors concluded that HyperProgressive Disease is more common with PD-1/PD-L1 inhibitors compared with chemotherapy, among previously treated patients with advanced NSCLC, and is also associated with high number of metastatic sites at baseline and poor survival. They added that the present study is the largest analysis exploring HPD to date and is the first conducted, in a dedicated NSCLC population, with a control cohort of chemotherapy-treated patients Hyperprogressive Disease in Patients With Advanced Non–Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. Ferrara R, Mezquita L, Texier M, et al. JAMA Oncol. Published online September 6, 2018. doi:10.1001/jamaoncol.2018.3676
Tumor Mutation Burden is a Predictive Biomarker for Response to Immune Checkpoint Inhibitors
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Patients with advanced NSCLC (Non-Small Cell Lung Cancer) often receive either platinum-doublet chemotherapy combination as first line therapy or KEYTRUDA® (Pembrolizumab) if the tumor PD-L1 expression is 50% or more. About 20-25% of patients benefit from immunotherapy. Other biomarkers besides PD-L1 are needed, to select appropriate patients for immunotherapy.
Tumor Mutational Burden (TMB) has recently emerged as a potential biomarker for immunotherapy with anti PD-1 antibodies. TMB can be measured using Next-Generation Sequencing (NGS) and is defined as the number of somatic, coding base substitutions and short insertions and deletions (indels) per megabase of genome examined. In a previously published trial (CheckMate 568), patients most likely to have a response to a combination of OPDIVO® (Nivolumab) plus YERVOY® (Ipilimumab), irrespective of tumor PD-L1 expression level in NSCLC, had a TMB of at least 10 mutations per megabase. This was the basis for CheckMate 227, which evaluated the efficacy of OPDIVO® and OPDIVO®-based regimens, as first line treatment in biomarker-selected groups of patients with advanced NSCLC.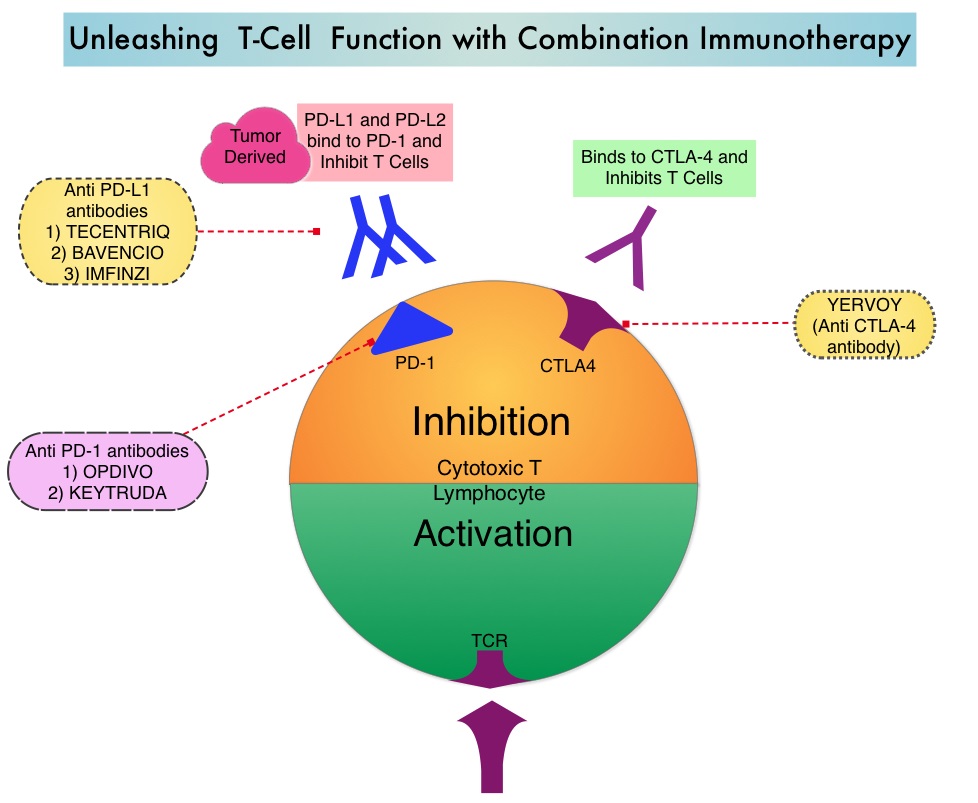
OPDIVO® is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, whereas YERVOY® is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4(Cytotoxic T-Lymphocyte Antigen 4, also known as CD152). Blocking the Immune checkpoint proteins unleashes the T cells, resulting in T cell proliferation, activation and a therapeutic response. The complementary mechanisms of action of OPDIVO® and YERVOY® combination resulted in greater efficacy in phase I trials, compared with OPDIVO® monotherapy.
CheckMate 227 is a three part, open-label, randomized, phase III trial, designed to compare different OPDIVO® -based regimens with chemotherapy in distinct patient populations. This study enrolled 1,739 patients with previously untreated Stage IV or recurrent NSCLC with no known sensitizing EGFR or ALK mutations and patients were randomized in a 1:1:1 ratio and the comparison was between either OPDIVO®, OPDIVO® plus YERVOY® or OPDIVO® plus platinum-doublet chemotherapy and platinum-doublet chemotherapy alone. Patients were stratified according to tumor histology and PD-L1 expression of 1% or more (positive) or less than 1% (negative). The study incorporated Tumor Mutational Burden (TMB) as a biomarker. This was determined by the FoundationOne CDx assay, an FDA approved test for molecular profiling, using the validated cutoff of TMB of 10 or more mutations/megabase as High, and less than 10 mutations/megabase as Low.
The authors in this publication reported data from part 1 of this study, which was a comparison between OPDIVO® plus YERVOY® versus chemotherapy, in patients with previously untreated Stage IV or recurrent NSCLC. In this comparison, 139 TMB-High patients were treated with OPDIVO® 3 mg/kg IV every 2 weeks plus YERVOY® 1 mg/kg IV every 6 weeks, whereas 160 TMB-High patients received chemotherapy, based on tumor histology. All treatments were continued until disease progression or unacceptable toxicity. Part 1 of the study had two Coprimary end points. One Coprimary end point was Progression Free Survival (PFS) with OPDIVO® plus YERVOY® versus chemotherapy in a patient population selected on the basis of TMB. The other Coprimary end point was Overall Survival (OS) with OPDIVO® plus YERVOY® versus chemotherapy in a patient population selected on the basis of the PD-L1 expression level.
It was noted that the PFS among patients with High TMB was significantly longer with OPDIVO® plus YERVOY®, compared with chemotherapy. The median PFS with the immunotherapy combination was 7.2 months compared to 5.5 months with chemotherapy (HR=0.58; P<0.001). This represented a 42% reduction in the risk of disease progression or death. The 1 year PFS more than tripled with combination immunotherapy at 42.6% versus 13.2% with chemotherapy. The Objective Response Rate (ORR) was 45.3% with immunotherapy combination and 26.9% with chemotherapy. The improved outcomes with OPDIVO® plus YERVOY® over chemotherapy was broadly consistent within all subgroups and was independent of tumor histology and PD-L1 expression. There was a clear trend toward improved survival with the immunotherapy combination although this data is immature. Grade 3 or 4 treatment related adverse events were 31.2% with immunotherapy combination and 36.1% with chemotherapy.
The authors concluded that this is the first phase III study to evaluate Tumor Mutational Burden as a predictive biomarker for immunotherapy as a coprimary endpoint. They added that these results highlight that Tumor Mutational Burden and PD-L1 are independent biomarkers and TMB is predictive of benefit with OPDIVO® plus YERVOY® irrespective of PD-L1 expression. TMB-High therefore is a new biomarker and represents a distinct subgroup of NSCLC. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. N Engl J Med 2018; 378:2093-2104
Late Breaking Abstract - ASCO 2018: Blood Test Demonstrates High Specificity for Detection of Early Stage Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Survival rates however are significantly higher when lung cancer is diagnosed early. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas, and 10% are Large cell carcinomas.
Although the U.S. Preventive Services Task Force (USPSTF) has recommended annual screening for lung cancer with Low-Dose Computed Tomography (LDCT) for individuals with significant smoking history, screening is vastly underutilized, with a screening rate of less than 2% among smokers eligible for screening. Screening for lung cancer using a peripheral blood sample may improve lung cancer screening rates. Analysis of cell-free DNA (cfDNA) from peripheral blood (Liquid Biopsy), is presently approved to select EGFR targeted therapies (cobas EGFR mutation test), in patients with advanced Non Small Cell Lung Cancer. However, the role of cell-free DNA analysis for early detection of lung cancer is not well established.
The Circulating Cell-Free Genome Atlas (CCGA) is a prospective, multi-center, observational study and is the largest study ever initiated, to develop a noninvasive, liquid biopsy assay for early cancer detection, based on cell-free DNA (cfDNA). This study has currently enrolled 10,012 of a planned 15,000 participants, including people with a recent cancer diagnosis and also a control group of individuals with no known malignancy (70% with cancer, 30% without cancer), across 141 sites in the United States and Canada. This report is one of the first pre-planned sub-studies from the CCGA, involving investigation of blood samples from 1,627 participants (878 patients with newly diagnosed untreated cancer including 127 patients with lung cancer and 749 controls - 580 controls and 169 technical assay controls ), across 20 tumor types and all clinical stages.
The cell-free DNA was isolated from the peripheral blood and analyzed using the following three sequencing methods that were designed to detect cancer-defining signals (mutations and other genomic changes), that could be utilized for early cancer detection.
Targeted sequencing to detect somatic (non-inherited) mutations, such as Single Nucleotide Variants and small insertions and/or deletions, in specific sections of the genome.
Whole-Genome Sequencing (WGS) to detect somatic gene copy number changes across the genome.
Whole-Genome Bisulfite Sequencing (WGBS) of cfDNA to detect abnormal patterns of cfDNA methylation (epigenetic changes)
In this initial sub-study, the authors explored the ability of the above three different assays to detect cancer in 127 people with stage I-IV lung cancer. It was noted that biologic signals suggesting lung cancer were detected and comparable across all assays, and the signal increased with cancer stage. At 98% specificity, the Targeted sequencing detected 51% of early-stage (stage I-IIIA) lung cancers and 89% of late-stage (stage IIIB-IV) lung cancers. Whole-Genome Sequencing detected 38% of early-stage cancers and 87% of late-stage cancers. Whole-Genome Bisulfite Sequencing had similar efficacy, detecting 41% of early stage lung cancers and 89% of late-stage cancers. Similar sensitivities were noted across all assays for adenocarcinoma, squamous cell and small cell lung cancer. False positive rates were low. Of the 580 control participants without cancer at study enrollment, less than 1% (five participants) had cancer-like signal across all three assays, of whom two were subsequently diagnosed with cancer. This highlights the potential for these assays to detect early stage cancers. The authors caution that a large proportion of cell-free DNA is derived from White Blood Cells (WBCs) and DNA mutations in the WBC population can also be generated by processes other than cancer such as clonal hematopoiesis during human aging. In this study, signal generated from the WBCs was subtracted resulting in a cleaner signal, only from tumor related variants.
It was concluded that based on the initial results from the CCGA study, it is possible to detect early-stage lung cancer, with a high degree of specificity, from a simple blood test, using genome sequencing. The authors plan to further optimize the assays and validate results in a larger group of people. Genome-wide sequencing for early stage lung cancer detection from plasma cell-free DNA (cfDNA): The Circulating Cancer Genome Atlas (CCGA) study. Oxnard GR, Maddala T, Hubbell E, et al. J Clin Oncol. 2018;36(suppl; abstr LBA8501)
Late Breaking Abstract - ASCO 2018: First Line TECENTRIQ® plus Chemotherapy in Advanced Squamous NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas, and 10% are Large cell carcinomas. Non Small Cell Lung Cancer patients with Squamous Cell histology have been a traditionally hard- to-treat, patient group, and less than 15% of patients with advanced Squamous NSCLC survive a year after diagnosis and less than 5% of patients survive for five years or longer. Immunotherapy is an accepted second line intervention after Platinum-based chemotherapy, in patients with advanced NSCLC, and is an approved first line therapy, for patients with high PD-L1 expressing tumors (50% or more).
TECENTRIQ® (Atezolizumab) is an anti PD-L1 monoclonal antibody, designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors and thus enabling the activation of T cells. TECENTRIQ® was approved by the FDA in October 2016 for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC) whose disease progressed during or following Platinum-containing chemotherapy. In this present publication, the authors studied the efficacy of TECENTRIQ® given along with combination chemotherapy, in patients with advanced Squamous NSCLC.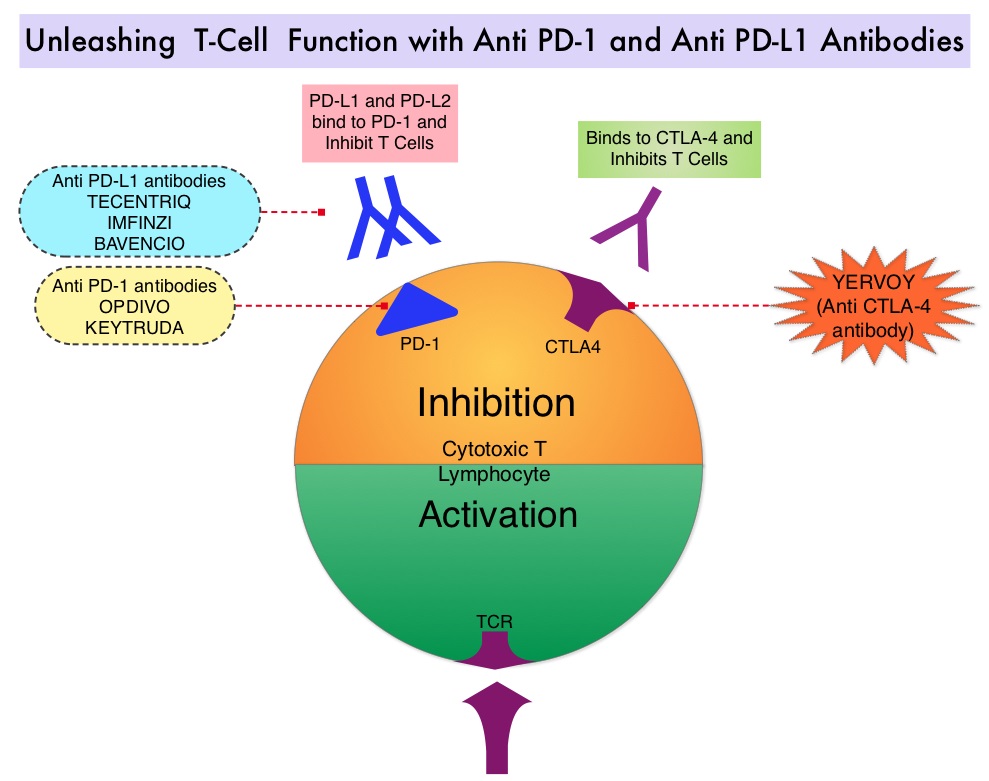
IMpower131 is a multicenter, open-label, phase III study, in which 1021 chemotherapy-naïve patients with stage IV Squamous NSCLC were randomly assigned in 1:1:1 ratio to receive TECENTRIQ® along with Carboplatin, and Paclitaxel (Group A, N=338), TECENTRIQ® along with Carboplatin, and ABRAXANE® (nab-paclitaxel) (Group B, N=343) and the control arm of Carboplatin and ABRAXANE® (Group C, N=340). Patients in Group A received TECENTRIQ® 1200 mg IV along with Carboplatin AUC 6 and TAXOL® (Paclitaxel) 200 mg/m2 IV, all drugs given on Day 1, every 21 days. Patients in Group B received TECENTRIQ® 1200 mg IV along with Carboplatin AUC 6 IV on Day 1 and ABRAXANE® 100mg/m2 IV on Days 1, 8, and 15 of each 21-day cycle. Patients in Group C (control group) received Carboplatin AUC 6 IV on Day 1 and ABRAXANE® 100mg/m2 IV on Days 1, 8, and 15 of each 21-day cycle. Patients received 4-6 cycles of this combination treatment and in Groups A and B, TECENTRIQ® alone was continued as long as there was a clinical benefit, without evidence of disease progression. Tumors were tested for PD-L1 expression, but patients were included in the study regardless of PD-L1 expression level. Patients with tumors demonstrating EGFR or ALK gene changes should have received molecularly targeted treatments before enrolling in this study. The co-Primary endpoints for this study were Progression Free Survival (PFS) and Overall Survival (OS). As per the study design, the current analysis compared the outcomes of patients in Group B with Group C. Outcomes data comparing Group A with Group C are not yet available.
At the time of primary analysis, with a median follow up of 17.1 months, the median PFS across all PD-L1 subgroups was 6.3 months with the addition of TECENTRIQ® to chemotherapy (Group B) versus 5.6 months in Group C, with chemotherapy alone (HR=0.71; P=0.0001). This represented a 29% reduction in the risk of disease progression or death, with the addition of TECENTRIQ® to chemotherapy. The 12-month PFS rates in Groups B and C were 24.7% versus 12.0%, respectively, suggesting a doubling of PFS benefit with the addition of TECENTRIQ® to chemotherapy. The PFS benefit was more pronounced in those with higher tumor PD-L1 expression. Overall Survival data are not yet mature. The most common side effects with the addition of TECENTRIQ® to chemotherapy included skin rash, colitis, and hypothyroidism.
The authors concluded that this is the first phase III trial of an immunotherapy-based treatment regimen, to demonstrate a significant improvement in Progression Free Survival, in advanced Squamous NSCLC. IMpower131: Primary PFS and safety analysis of a randomized phase III study of atezolizumab + carboplatin + paclitaxel or nab-paclitaxel vs carboplatin + nab-paclitaxel as 1L therapy in advanced squamous NSCLC. Jotte RM, Cappuzzo F, Vynnychenko I, et al. J Clin Oncol 36, 2018 (suppl; abstr LBA9000)
Late Breaking Abstract - ASCO 2018: First-Line KEYTRUDA® Superior to Chemotherapy in NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®. The FDA approved KEYTRUDA® for the first-line treatment of advanced NSCLC with high PD-L1 expression (Tumor Proportion Score of 50% or more), as well as in combination with Pemetrexed and Carboplatin, as first-line treatment of patients with metastatic nonsquamous NSCLC and for previously treated advanced NSCLC with a PD-L1 Tumor Proportion Score of 1% or more. Currently, KEYTRUDA® is the only FDA approved immunotherapy for initial treatment of NSCLC as monotherapy (KEYNOTE-024) or in combination with chemotherapy. In KEYNOTE-024, KEYTRUDA® significantly improved Progression Free Survival and Overall Survival compared to chemotherapy, as first-line treatment for metastatic NSCLC, without targetable mutations and PD-L1 TPS of 50% or more. KEYNOTE-042 trial evaluated the benefit of KEYTRUDA® in patients whose tumors had a much lower level of PD-L1 expression (TPS of 1% or higher).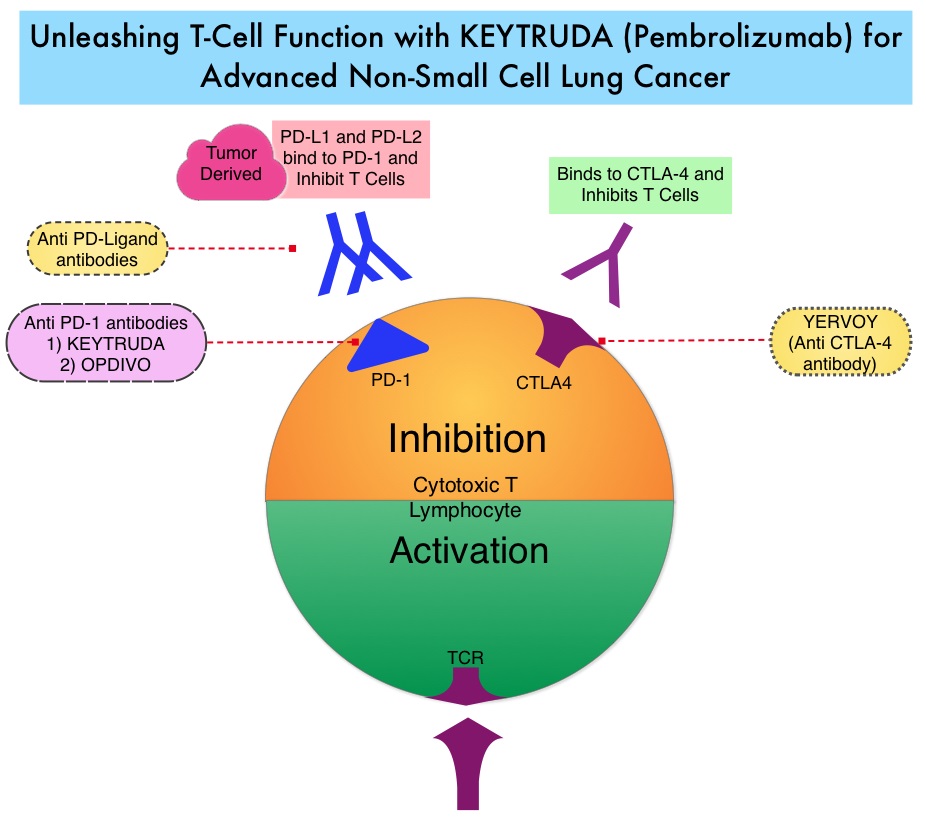
KEYNOTE-042 is a large, international, multicenter, randomized phase III trial in which 1274 patients with untreated locally advanced or metastatic NSCLC were randomly assigned to KEYTRUDA® or chemotherapy with Paclitaxel plus Carboplatin or Pemetrexed plus Carboplatin. In this study, both squamous and non-squamous cancers with PD-L1 Tumor Proportion Score (TPS) of 1% or more were included, but tumors with sensitizing Epidermal Growth Factor Receptor (EGFR) or Anaplastic Lymphoma Kinase (ALK) mutations cancers with genetic changes, that could be treated with targeted therapies such as EGFR and ALK inhibitors, were excluded. Eligible patients were randomly assigned in a 1:1 to receive either KEYTRUDA® 200 mg IV every 3 weeks for up to 35 cycles or investigator’s choice of up to 6 cycles of chemotherapy with Paclitaxel plus Carboplatin or Pemetrexed plus Carboplatin, with optional Pemetrexed maintenance for nonsquamous NSCLC. Patients were divided into 3 treatment groups based on their PD-L1 Tumor Proportion Score (TPS): TPS 50% or more (N=599), TPS 20% or more (N=818), and TPS 1% or more (N=1274). Each PD-L1 expression group had equal numbers of patients receiving KEYTRUDA® and chemotherapy. The Primary end points were Overall Survival (OS) in patients with TPS 50% or more, 20% or more, and 1% or more.
At a median follow up of 12.8 months, 13.7% of patients were still receiving KEYTRUDA® compared with 4.9% on Pemetrexed maintenance therapy. It was noted that KEYTRUDA® was significantly superior to chemotherapy in all PD-L1 expression subsets. In patients with a PD-L1 TPS 50% or more, the median OS with KEYTRUDA® was 20 months versus 12.2 months for chemotherapy (HR=0.69, P=0.0003), for patients with PD-L1 TPS 20% or more, the median OS was 17.7 months versus 13 months respectively (HR=0.77, P=0.002), and for those with PD-L1 TPS 1% or more, the median OS was 16.7 months versus 12.1 months respectively (HR=0.81, P = 0.0018). The Response Rates (RR) were also higher among patients who received KEYTRUDA®, with RR of 39.5% for KEYTRUDA® versus 32% for chemotherapy in patients with a TPS 50% or more, 33.4% and 28.9% respectively in patients with TPS 20% or more and 27.3% and 26.5%, respectively, in patients with TPS of 1% or more. The duration of response was also superior with KEYTRUDA® in all three PD-L1 subgroups compared to chemotherapy (20.2 months versus 8-11 months). Patients receiving KEYTRUDA® experienced fewer severe Adverse Events, compared with chemotherapy (17.8% versus 41%).
The authors concluded that this is the largest clinical trial of KEYTRUDA® as a stand-alone therapy, and is the first study with a Primary end point of OS to demonstrate superiority of KEYTRUDA® over platinum-based chemotherapy, in patients with previously untreated advanced/metastatic NSCLC, without sensitizing EGFR or ALK alterations and a PD-L1 TPS of 1% or more. These data confirmed the benefit of KEYTRUDA® monotherapy as a standard first-line treatment, for PD-L1-expressing advanced/metastatic NSCLC. Pembrolizumab (pembro) versus platinum-based chemotherapy (chemo) as first-line therapy for advanced/metastatic NSCLC with a PD-L1 tumor proportion score (TPS) ≥ 1%: Open-label, phase 3 KEYNOTE-042 study. Lopes G, Wu Y-L, Kudaba I, et al. J Clin Oncol 36, 2018 (suppl; abstr LBA4)
Lung Immune Prognostic Index (LIPI) Identifies Advanced Non Small Cell Lung Cancer Patients Unlikely to Benefit from Treatment with Immune Checkpoint Inhibitors
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas.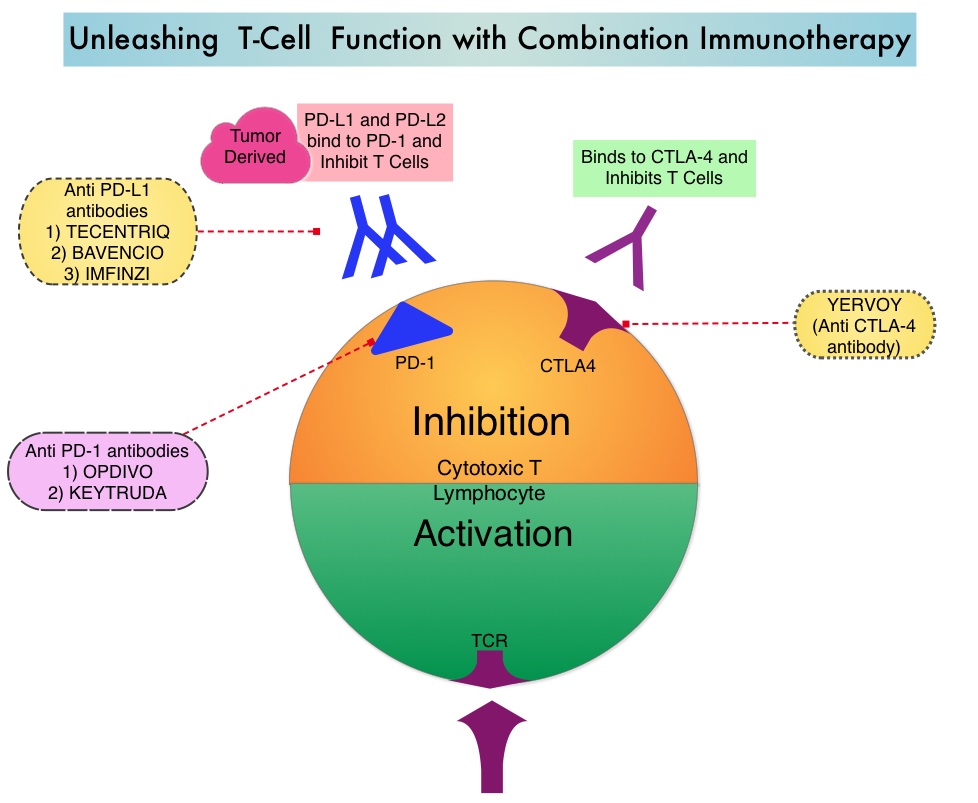
Immunotherapy with PD-1/PD-L1 (Programmed Death-1/Programmed Death-Ligand 1) inhibitors, also called Immune Checkpoint Inhibitors (ICIs), has changed the treatment paradigm for patients with advanced NSCLC. In previously treated patients with NSCLC, the Overall Response Rates (ORR) with single agent Immune Checkpoint Inhibitors (ICIs) range from 14-20%, with median Overall Survival (OS) of 10 to 12 months. In those with PD-L1 expression of 50% or more by ImmunoHistoChemical (IHC) analysis, the ORR can reach up to 30% with a median OS of 20 months. However, in patients with negative or weak PD-L1 expression (1%-49% positive tumor cells), who account for approximately two thirds of the NSCLC population, the response rates range from 8-19% with a median OS slightly below 10 months. Even among those with tumors expressing PD-L1 expression of 50% or more, not all patients benefit from Immunotherapy with ICIs. Therefore identifying biomarkers for patients likely to respond to ICI therapy, and predicting resistance is important and relevant in selecting the appropriate patients for treatment with ICIs.
There is growing evidence on the role of inflammation in cancer biology and systemic inflammatory response may have prognostic significance in different cancer types. Inflammatory process in various cancers imparts immunoresistance to ICIs, by activating oncogenic signaling pathways, there by promoting cancer growth and dissemination, with resulting poor outcomes. Derived Neutrophil-to-Lymphocyte ratio (dNLR) and serum Lactate DeHydrogenase (LDH) level have been investigated as potential inflammatory biomarkers in patients with cancer. The dNLR is calculated using a formula dNLR= Absolute Neutrophil Count/(White Blood Count - Absolute Neutrophil Count). These ratios are simple and easy to calculate from routine blood tests. Both these biomarkers have been correlated with Immune Checkpoint Inhibitor outcomes, in patients with melanoma. In two large studies involving patients with advanced melanoma treated with Ipilimumab and Pembrolizumab, dNLR of 3 or more and LDH of at least 2.5 times Upper Limit of Normal (ULN), reflected a pro-inflammatory status and resulted in poor outcomes.
Based on this important finding in malignant melanoma, the authors conducted a multicenter, retrospective study to determine whether combining the two factors - pretreatment dNLR and LDH (Lung Immune Prognostic Index-LIPI), was associated with resistance to ICIs in patients with advanced NSCLC. In this study, LIPI was developed on the basis of dNLR (derived Neutrophil-to-Lymphocyte Ratio) of greater than 3 and LDH greater than Upper Limit of Normal (ULN). LIPI was used to stratify patients with NSCLC into 3 groups (Good= 0 factors; Intermediate= 1 of 2 factors, Poor= 2 factors). The pooled cohort treated with ICIs included 466 patients with advanced NSCLC of whom 161 patients were treated at a single institution, to test the potential of the biomarkers score (test cohort) and the hypothesis was then validated in a larger multicentric validation cohort of 305 patients treated at 8 European academic centers. To determine whether the LIPI is specific to Immune Checkpoint Inhibitors (ICIs), a control cohort of 162 patients with advanced NSCLC, treated exclusively with chemotherapy, were also evaluated for LIPI. The median patient age was 62 years, 58% had Adenocarcinoma and 34% had Squamous histology, 74% had PD-L1 expression of at least 1% by IHC analysis and 26% had negative results. Median follow up was 12 months. The Primary end point was Overall Survival (OS) and Secondary end points included Progression Free Survival (PFS) and Disease Control Rate (DCR).
It was noted that in the test cohort, median PFS and OS were 3 and 10 months, respectively which is consistent with prior reports in patients with NSCLC, treated with PD-1 inhibitors in second or later lines. A dNLR greater than 3 and LDH greater than ULN were independently associated with OS. The median OS for Poor, Intermediate, and Good LIPI was 3 months, 10 months and 34 months respectively, and median PFS was 2 months, 3.7 months and 6.3 months respectively (P<0.001). This suggested that the population with a Poor (high) LIPI were more likely to have progressive disease as their best response to immunotherapy and had both shorter PFS and OS, compared to those with an Intermediate or Good (low) LIPI. Disease Control Rate (Complete plus Partial Response plus Stable disease) was also correlated with dNLR greater than 3 and LDH greater than ULN. These results were reproducible in the ICI validation cohort for OS, PFS, and DCR. LIPI however was not associated with outcome in patients treated with chemotherapy only, providing support that it might be a predictor of benefit from Immune Checkpoint Inhibitors (ICIs).
It was concluded that pretreatment LIPI, combining derived Neutrophil-to-Lymphocyte ratio (dNLR) greater than 3 and serum LDH level greater than Upper Limit of Normal, correlated with worse outcomes for Immune Checkpoint Inhibitors (ICIs). The authors suggested that this is the first study to explore LIPI in NSCLC and can serve as a potentially useful tool when selecting patients for treatment with Immune Checkpoint Inhibitors, and LIPI might be useful for identifying patients unlikely to benefit from treatment with an ICI. Association of the Lung Immune Prognostic Index With Immune Checkpoint Inhibitor Outcomes in Patients With Advanced Non–Small Cell Lung Cancer. Mezquita L, Auclin E, Ferrara R, et al. JAMA Oncol. 2018;4:351-357
KEYTRUDA® Plus Standard Chemotherapy Improves Overall Survival in Newly Diagnosed Metastatic Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas.
KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®. The FDA approved KEYTRUDA® for the first-line treatment of advanced NSCLC with high PD-L1 expression (Tumor Proportion Score of 50% or more), and for previously treated advanced NSCLC with a PD-L1 Tumor Proportion Score of 1% or more. This present study provides convincing phase III evidence to support the use of triplet therapy in NSCLC.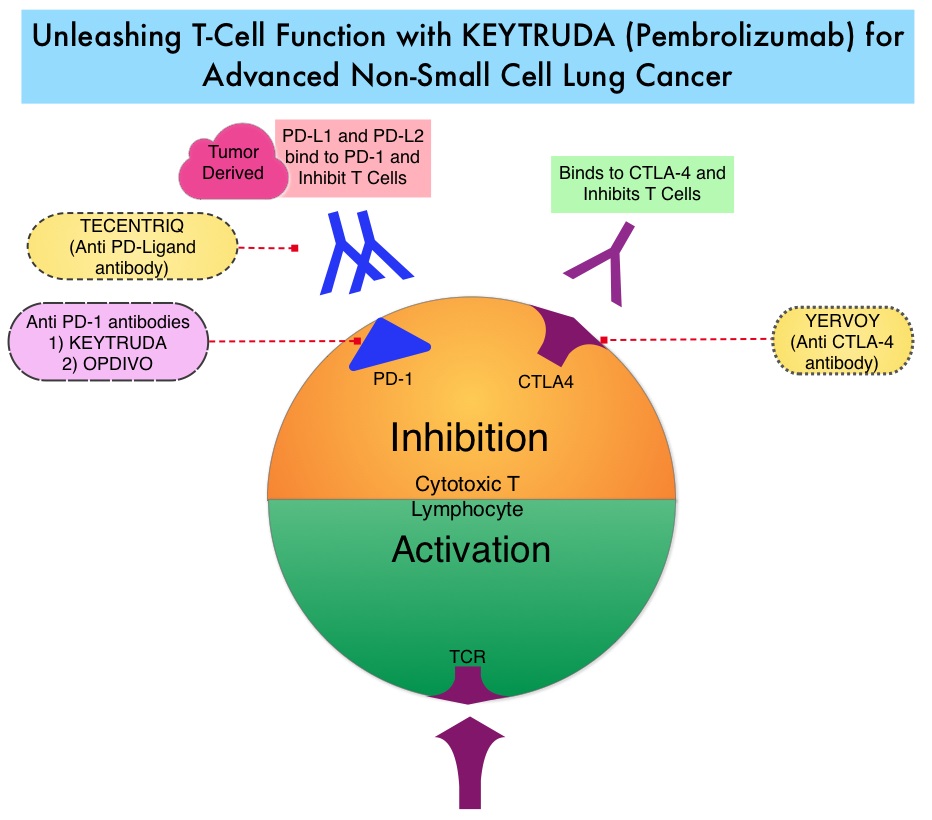
KEYNOTE-189 is a double-blind, phase III trial in which 616 patients with untreated stage IV non-squamous NSCLC, without sensitizing EGFR or ALK mutations, were randomly assigned in a 2:1 ratio to receive treatment with four cycles of KEYTRUDA®/Pemetrexed/Carboplatin or placebo plus the same chemotherapy. Patients received either KEYTRUDA® 200mg or saline placebo, both administered IV every 3 weeks for up to 35 cycles. All the patients received four cycles of the investigator’s choice of Cisplatin 75 mg/m2 IV or Carboplatin AUC 5 along with Pemetrexed 500 mg/m2, all administered IV every 3 weeks, followed by maintenance Pemetrexed 500 mg/m2 every 3 weeks. Patients in the placebo combination group were allowed to crossover to KEYTRUDA® monotherapy upon disease progression. Patients with symptomatic brain metastasis were excluded and patients were stratified according to PD-L1 expression (Tumor Proportion Score, 1% or more versus less than 1%), choice of platinum-based drug (Cisplatin versus Carboplatin), and smoking history. Both treatment groups were well balanced and about 17% had brain metastasis and one-third were untreated. A PD-L1 Tumor Proportion Score of 1% or more was reported in 63% of the patients, Carboplatin was the preferred platinum-based drug in 72% of the patients, and 88% of the patients were current or former smokers. The co-Primary end points were Overall Survival (OS) and Progression Free Survival (PFS).
After a median follow-up of 10.5 months, the estimated rate of Overall Survival (OS) at 12 months was 69.2% in the KEYTRUDA®-combination group versus 49.4% in the placebo-combination group. The median OS was not reached in the KEYTRUDA®-combination group and was 11.3 months in the placebo-combination group (HR=0.49; P<0.001). This represented a 51% reduction in the risk of death in the KEYTRUDA®-combination group. The OS benefit was seen across all PD-L1 subgroups including those with a PD-L1 Tumor Proportion Score of less than 1%. However, the greatest benefit was noted in the group expressing high levels of PD-L1 with Tumor Proportion Score 50% or more (12-month OS rate of 73% versus 48.1%; HR=0.42). The median Progression Free Survival was 8.8 months in the KEYTRUDA®-combination group and 4.9 months in the placebo-combination group (HR=0.52; P<0.001). This represented a 48% reduction in the risk of disease progression. The Objective Response Rate (ORR) was 47.6% in the KEYTRUDA®-combination group and 18.9% in the placebo-combination group (P<0.001) and ORR was highest among those with a PD-L1 Tumor Proportion Score of 50% or more (61.4% vs 22.9%). There was no significant difference in grade 3 adverse events in the two treatment groups.
It was concluded that in patients with previously untreated metastatic non-squamous NSCLC without EGFR or ALK mutations, the addition of KEYTRUDA® to standard chemotherapy of Pemetrexed and a platinum-based drug resulted in significantly longer Overall Survival and Progression Free Survival than chemotherapy alone, regardless of PD-L1 status and should therefore be considered as a new standard of care, in the front-line setting. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. Gandhi L, Rodriguez-Abreu D, Gadgeel S, et al. for the KEYNOTE-189 Investigators. April 16, 2018 DOI: 10.1056/NEJMoa1801005
FDA Approves TAGRISSO® for First-Line Treatment of Metastatic NSCLC
SUMMARY: The FDA on April 19, 2018, approved TAGRISSO® (Osimertinib) for the first-line treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 L858R mutations, as detected by an FDA-approved test.
Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Approximately 10-15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either exon 19 deletions or L858R point mutations in exon 21. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients. Put another way, T790M is not relevant in about 40% of patients whose disease progression may be related to other mechanisms.
EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50-60% of patients. Put another way, T790M is not relevant in about 40% of patients whose disease progression may be related to other mechanisms.
TAGRISSO® is a third-generation Epidermal Growth Factor Receptor (EGFR) TKI presently approved by the FDA, for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, who had progressed on prior systemic therapy, including an EGFR-TKI. Previously published studies had suggested that TAGRISSO® may also be effective as initial therapy for EGFR mutation-positive advanced NSCLC.
The recent new indication for TAGRISSO® was based on FLAURA, which is a randomized, double blind, phase III clinical trial, conducted to compare the efficacy and safety of first line TAGRISSO® to TARCEVA® or IRESSA® (which are considered standard first line therapies), in NSCLC patients with activating mutations EGFR exon 19 deletions or L858R substitution mutation on exon 21. This study randomized 556 advanced NSCLC treatment naïve patients, with EGFR exon 19 or 21 mutations in a 1:1 ratio, to TAGRISSO® 80 mg orally once daily (N=279) or Standard of Care EGFR-TKI, IRESSA® 250 mg or TARCEVA® 150 mg, orally once daily (N=277). Patients were stratified by mutation status (exon 19 vs 21 mutations) and race (Asian vs non-Asian). Patients with CNS metastases who were neurologically stable, were allowed in this study. The Primary endpoint was Progression Free Survival (PFS).
The median PFS was 18.9 months with TAGRISSO® compared to 10.2 months for the standard therapy (HR=0.46; P<0.001), suggesting a 54% reduction in the risk of disease progression, compared with Standard of Care. TAGRISSO® extended the median Time To Progression by about 9 months. This PFS benefit was consistent across all subgroups of patients, including those with and without CNS metastases at study entry. The Objective Response Rate (ORR) with TAGRISSO® was 80% compared with 76% for TARCEVA® and IRESSA®. The median Duration of Response with TAGRISSO® was 17.2 months versus 8.5 months in the comparator arm. The median Overall Survival was not reached. Grade 3 and 4 toxicities were lower for TAGRISSO® (34%) compared with 45% for TARCEVA® and IRESSA®. Toxicities led to treatment discontinuation for 13% and 18% of patients in the TAGRISSO® and comparator groups, respectively.
It was concluded that TAGRISSO® demonstrated superior efficacy, with a near doubling in median Progression Free Survival, and better tolerability, compared to the Standard of Care, when given as first-line therapy, for patients with advanced EGFR mutation positive NSCLC. Studies are underway, assessing treatments, following resistance to TAGRISSO®.
Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. Soria J-C, Ohe Y, Vansteenkiste J, et al. for the FLAURA Investigators. N Engl J Med 2018; 378:113-125
Frontline TECENTRIQ® along with AVASTIN® and Chemotherapy Improves Survival in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. Immunotherapy is an accepted second line intervention after platinum-based chemotherapy in patients with advanced NSCLC, and is an approved first line therapy, for patients with high PD-L1 expressing tumors (50% or more). Further, immunotherapy with KEYTRUDA® (Pembrolizumab), in combination with chemotherapy, has been approved for first line treatment of patients with advanced non-squamous NSCLC, irrespective of PD-L1 expression.
TECENTRIQ® (Atezolizumab) is an anti-PDL1 monoclonal antibody, designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors and thus enabling the activation of T cells. TECENTRIQ® was approved by the FDA in October 2016 for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC) whose disease progressed during or following Platinum-containing chemotherapy. AVASTIN® (Bevacizumab) is a biologic antiangiogenic antibody, directed against Vascular Endothelial Growth Factor (VEGF), and prevents the interaction of VEGF to its receptors (Flt-1 and KDR) on the surface of endothelial cells. The interaction of VEGF with its receptors has been shown to result in endothelial cell proliferation and new blood vessel formation. Combining TECENTRIQ® and AVASTIN® is supported by the following scientific rationale. AVASTIN® in addition to its established anti-angiogenic effects, may further enhance the ability of TECENTRIQ® to restore anti-cancer immunity, by inhibiting VEGF-related immunosuppression, promoting T-cell tumor infiltration and enabling priming and activation of T-cell responses against tumor antigens.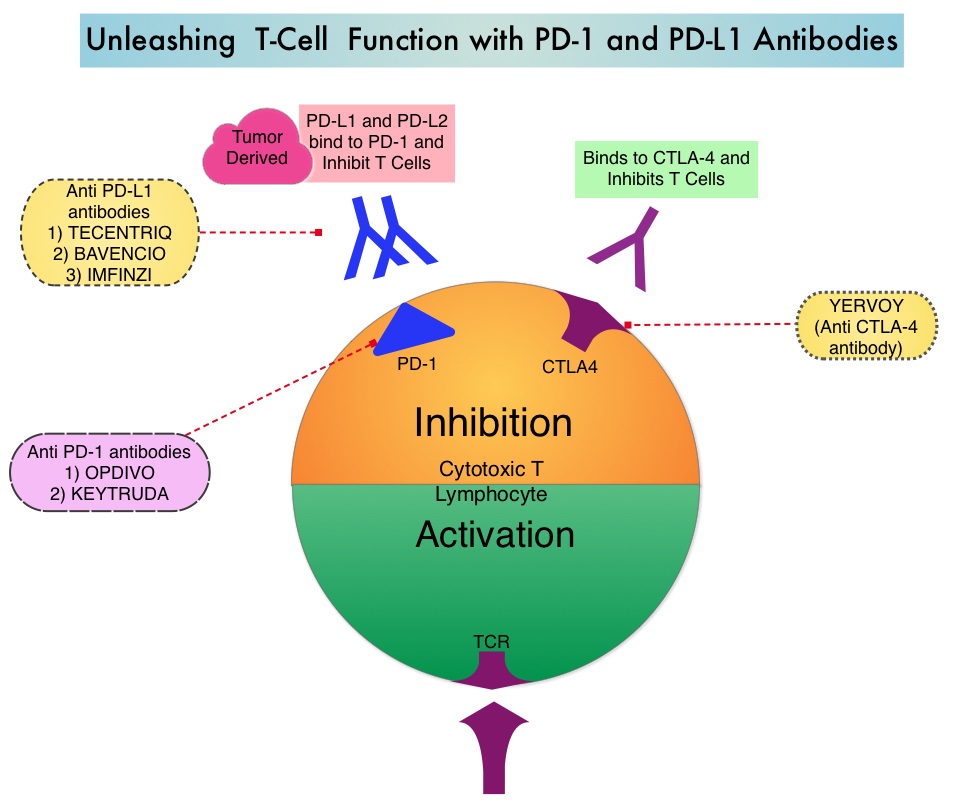
IMpower150 is a multicenter, open-label, randomized, phase III study, conducted to evaluate the efficacy and safety of TECENTRIQ® in combination with Carboplatin and Paclitaxel with or without AVASTIN®, in patients with stage IV, treatment naïve, non-squamous NSCLC. This study enrolled 1,202 patients, who were randomized (1:1:1) to receive either TECENTRIQ® along with Carboplatin and Paclitaxel (Group A), TECENTRIQ® and AVASTIN® along with Carboplatin and Paclitaxel (Group B), or AVASTIN® plus Carboplatin and Paclitaxel (Group C - control arm). During the treatment-induction phase, patients in Group A received TECENTRIQ® 1200 mg IV along with Carboplatin AUC 6 and Paclitaxel 200mg/m2 IV on Day 1 of a 3-week treatment cycle for 4 or 6 cycles. Following the induction phase, patients received maintenance treatment with TECENTRIQ® on the same dose schedule until disease progression. Patients in Group B received AVASTIN® 15 mg/kg IV, along with TECENTRIQ®, Carboplatin and Paclitaxel IV, Day 1 of a 3-week treatment cycle for 4 or 6 cycles followed by maintenance treatment with the TECENTRIQ® and AVASTIN® until disease progression. Patients in the control Group C received AVASTIN® plus Carboplatin and Paclitaxel every 3 weeks for 4 or 6 cycles followed by AVASTIN® maintenance treatment until disease progression. Patients with tumors demonstrating ALK and EGFR mutations were excluded from the primary Intention-To-Treat (ITT) analysis. Patients were also tested for a tumor T-effector gene expression signature (based on phase II trial finding of prolonged Overall Survival in patients with high gene expression signature levels, treated with TECENTRIQ®). The median age was 63 years and the minimum follow up at the time of the analysis was 9.5 months. For the interim analysis, the study was only designed to compare Groups B and C. The co-Primary endpoints were Progression Free Survival (PFS) and Overall Survival in the Intention-to-Treat (ITT) population comparing patients in Group B and C. These end points were also evaluated in subgroup of people who had a specific biomarker (T-effector gene signature expression).
It was noted that at this interim analysis, the combination of TECENTRIQ® and AVASTIN® plus Carboplatin and Paclitaxel, significantly improved PFS and reduced the risk of disease worsening or death by 38% (HR=0.62; P<0.0001), compared to AVASTIN® plus Carboplatin and Paclitaxel alone. This PFS benefit was observed across key subgroups, regardless of PD-L1 expression status, including PD-L1–negative patients (HR 0.77). Further, the median PFS in the population of patients with defined expression of a T-effector gene signature expression in the tumor tissue, was 11.3 months versus 6.8 months (HR 0.51; P<0.0001). Roche on March 26, 2018 announced that the IMpower150 study met its co-primary endpoint of Overall Survival as well. Details will soon become available.
It was concluded that combining chemotherapy with immunotherapy and antiangiogenic agents significantly improved PFS as well as Overall Survival, in patients with treatment naïve, advanced non-squamous NSCLC. This strategy can completely eliminate the need for patient selection based on a particular biomarker, and could benefit larger number of patients with advanced NSCLC. Reck M. Primary PFS and safety analyses of a randomized Phase III study of carboplatin + paclitaxel +/− bevacizumab, with or without atezolizumab in 1L non-squamous metastatic NSCLC (IMpower150). Annals of Oncology, 2017;28(11). Abstract LBA1_PR. https://www.roche.com/media/store/releases/med-cor-2018-03-26.htm
Long Term Survival Outcomes with OPDIVO® in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Under normal circumstances, Immune checkpoints or gate keepers inhibit intense immune responses by switching off the T cells of the immune system. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies have been developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By doing so, T cells are unleashed, resulting in T cell proliferation, activation and a therapeutic response.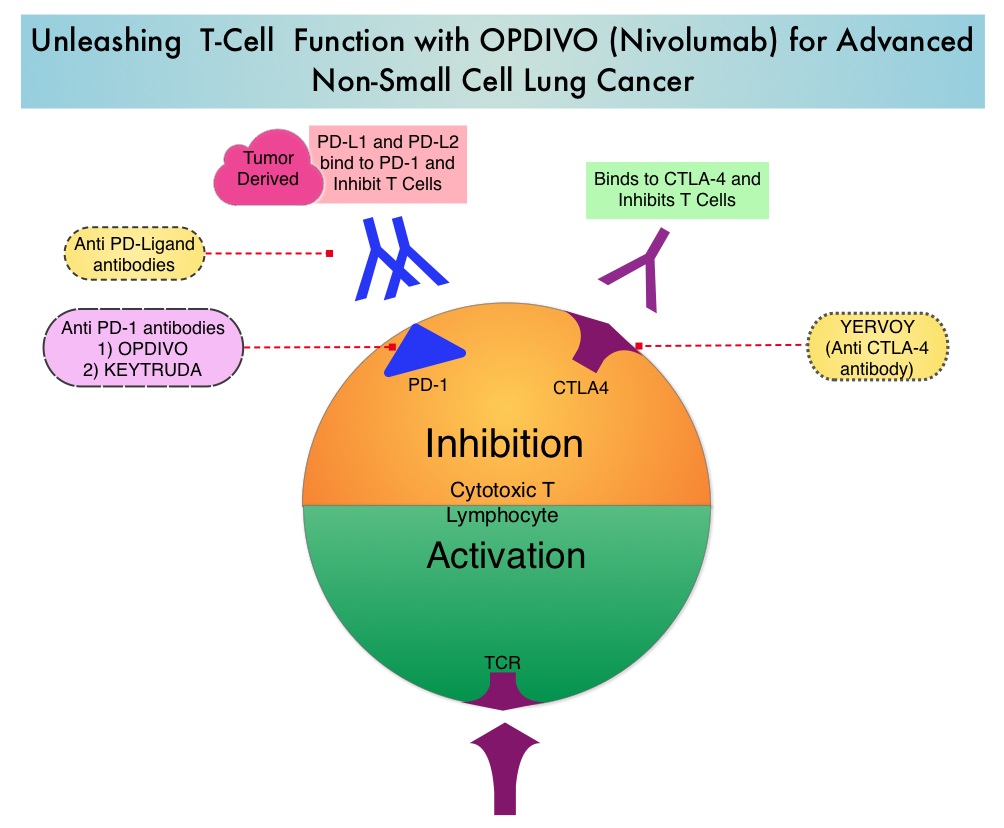
OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. OPDIVO® significantly prolonged Overall Survival (OS) and had a favorable toxicity profile, when compared to TAXOTERE® (Docetaxel), in two open-label, randomized, phase III trials, among patients with advanced squamous (CheckMate 017) or non-squamous (CheckMate 057) NSCLC, who had disease progression during or after platinum-based chemotherapy. This data led to the approval of OPDIVO® in previously treated advanced NSCLC.
There is however limited data on long term efficacy and safety for immune checkpoint inhibitors in patients with NSCLC when compared with chemotherapy. The authors in this publication reported updated efficacy and safety data for OPDIVO® in patients with advanced NSCLC from the CheckMate 017 and CheckMate 057 trials, with a minimum follow up of 2 years in all patients. Patients in both trials had stage IIIB or IV disease and had disease progression during or after platinum-based chemotherapy. In CheckMate 017 study, 272 patients with metastatic squamous NSCLC were randomized to receive OPDIVO® (Nivolumab) 3 mg/kg IV every 2 weeks (N=135) or TAXOTERE® (Docetaxel) 75 mg/m2 IV every 3 weeks (N=137). In CheckMate 057 trial, 582 patients were randomized to receive OPDIVO® 3 mg/kg IV every 2 weeks (N=292) or TAXOTERE® 75 mg/m2 IV every 3 weeks (N=290). Treatment was continued until disease progression or unacceptable toxicity. The minimum follow up for survival was 24.2 months.
The Overall Survival rate at 2 years with OPDIVO® versus TAXOTERE® were 23% vs 8% among squamous NSCLC patients and 29% vs 16% among non-squamous NSCLC patients. The relative reductions in the risk of death with OPDIVO® versus TAXOTERE® was similar to what was reported in the primary analyses. As was reported at the time of the primary analysis of these two trials, the superiority of OPDIVO® over TAXOTERE® was independent of Programmed cell Death Ligand 1 (PD-L1) expression in squamous NSCLC, whereas among patients with non-squamous histology, the benefit with OPDIVO® was greater in those with higher levels of PD-L1 expression, but patients with PD-L1 expression of less than 1% benefited as well.
In the pooled analysis of both trials, the median OS was 11.1 months with OPDIVO® versus 8.1 months with TAXOTERE® (HR=0.72). The relative reduction in the risk of death with OPDIVO® was 28%. Higher PD-L1 expression levels was associated with greater OS benefit with OPDIVO® (HR=0.42 in patients with 50% or more PD-L1 expression). Survival benefit however was still observed in patients with 1% or less PD-L1 expression (HR=0.78). Durable responses were noted with OPDIVO® among 37% of the confirmed responders with squamous NSCLC and 34% of confirmed responders with non-squamous NSCLC. These patients had ongoing responses after a minimum follow up of 2 years whereas no patient in the TAXOTERE® treated group had an ongoing response. The rates of treatment-related adverse events of any grade were lower with OPDIVO® compared to TAXOTERE® (any grade: 68% vs 88%; grade 3-4: 10% vs 55%, respectively).
The authors concluded that OPDIVO® provides long term clinical benefit and has a favorable tolerability profile compared with TAXOTERE®, in previously treated patients with advanced NSCLC. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non–Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). Horn L, Spigel DR, Vokes EE, et al. J Clin Oncol 2017;35:3924-3933
Molecular Testing in Lung Cancer - Guideline Update
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology convened an expert panel in 2013 and had published evidence-based guideline to set standards for the molecular analysis of lung cancers and to guide treatment decisions with targeted therapies. With the availability of new medical information and technological advances, this expert panel which comprised of pathologists, oncologists, pulmonologists, and laboratory scientists, issued an evidence based update which included 18 new recommendations, along with 3 updated recommendations from the 2013 guideline, asking 5 key questions.
Key Question 1: Which new genes should be tested for lung cancer patients?
a) ROS1 testing must be performed on all lung adenocarcinoma patients, irrespective of clinical characteristics.
b) ROS1 ImmunoHistoChemistry (IHC) may be used as a screening test in lung adenocarcinoma patients; however, positive ROS1 IHC results should be confirmed by a molecular or cytogenetic method.
c) BRAF, RET, ERBB2 (HER2), KRAS and MET molecular testing are currently not indicated as a routine stand-alone assay, outside the context of a clinical trial. It is appropriate to include molecular testing for these genes, as part of larger testing panels performed either initially or when routine EGFR, ALK, and ROS1 testing are negative.
Key Question 2: What methods should be used to perform molecular testing?
a) ImmunoHistoChemistry (IHC) is an equivalent alternative to Fluorescence In Situ Hybridization (FISH) for ALK testing.
b) Multiplexed genetic sequencing panels are preferred over multiple single-gene tests, to identify other treatment options beyond EGFR, ALK, and ROS1.
c) Laboratories should ensure test results that are unexpected, discordant, equivocal or otherwise of low confidence, are confirmed or resolved, using an alternative method or sample.
Key Question 3: Is molecular testing appropriate for lung cancers that do not have an adenocarcinoma component?
a) Physicians may use molecular biomarker testing in tumors with histologies other than adenocarcinoma when clinical features indicate a higher probability of an oncogenic driver.
Key Question 4: What testing is indicated for patients with targetable mutations who have relapsed on targeted therapy?
a) In lung adenocarcinoma patients who harbor sensitizing EGFR mutations and have progressed after treatment with an EGFR-targeted TKI, physicians must use EGFR T790M mutational testing when selecting patients for third-generation EGFR-targeted therapy.
b) Laboratories testing for EGFR T790M mutation in patients with secondary clinical resistance to EGFR-targeted kinase inhibitors should deploy assays capable of detecting EGFR T790M mutations in as little as 5% of viable cells.
c) There is currently insufficient evidence to support a recommendation for or against routine testing for ALK mutational status for lung adenocarcinoma patients with sensitizing ALK mutations, who have progressed after treatment with an ALK-targeted Tyrosine Kinase Inhibitor (TKI).
Key Question 5: What is the role of testing for circulating cell-free DNA for lung cancer patients?
a) There is currently insufficient evidence to support the use of circulating cfDNA molecular methods for the diagnosis of primary lung adenocarcinoma.
b) In some clinical settings in which tissue is limited and/or insufficient for molecular testing, physicians may use a cfDNA assay to identify EGFR mutations.
c) Physicians may use cfDNA methods to identify EGFR T790M mutations in lung adenocarcinoma patients with progression or secondary clinical resistance to EGFR-targeted TKI; testing of the tumor sample is recommended if the plasma result is negative.
d) There is currently insufficient evidence to support the use of circulating tumor cell molecular analysis for the diagnosis of primary lung adenocarcinoma, the identification of EGFR or other mutations, or the identification of EGFR T790M mutations at the time of EGFR TKI resistance.
2013 Statements VERSUS 2017 Statements
a) 2013 - Cytologic samples are also suitable for EGFR and ALK testing, with cell blocks being preferred over smear preparations VERSUS 2017 - Pathologists may use either cell blocks or other cytologic preparations as suitable specimens for lung cancer biomarker molecular testing.
b) 2013 - Laboratories should use EGFR test methods that are able to detect mutations in specimens with at least 50% cancer cell content, although laboratories are strongly encouraged to use (or have available at an external reference laboratory) more sensitive tests that are able to detect mutations in specimens with as little as 10% cancer cells VERSUS 2017 - Laboratories should use, or have available at an external reference laboratory, clinical lung cancer biomarker molecular testing assays that are able to detect molecular alterations in specimens with as little as 20% cancer cells.
c) 2013 - IHC for total EGFR is not recommended for selection of EGFR TKI therapy VERSUS 2017 - It is strongly recommended that laboratories should not use total EGFR expression by IHC testing to select patients for EGFR-targeted TKI therapy.
Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Lindeman NI, Cagle PT, Aisner DL et al. [published online January 22,2018]. Arch Pathol Lab Med . doi: 10.5858/arpa.2017-0388-CP
KEYTRUDA® Doubles Overall Survival Compared with Chemotherapy in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers.
KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor (immune checkpoint protein) and blocks its interaction with ligands PD-L1 and PD-L2. This leads to the undoing of the PD-1 pathway-mediated inhibition of the immune response and the tumor-specific effector T cells are unleashed. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced Non Small Cell Lung Cancer (NSCLC) have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.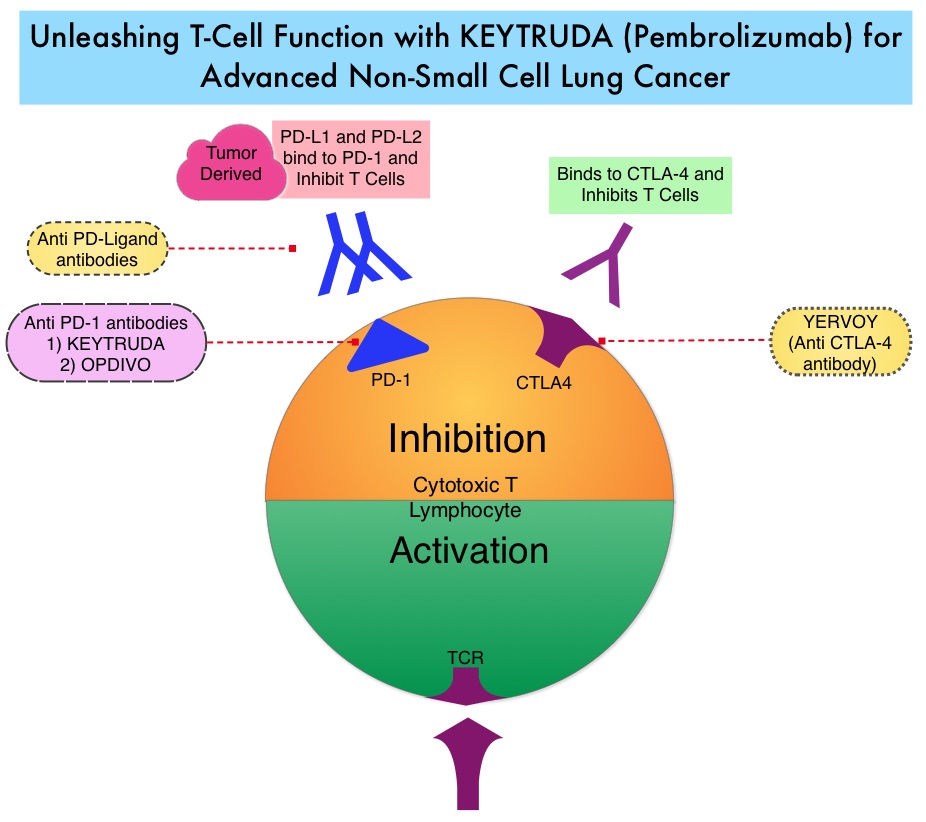
KEYNOTE-024 is an open-label, randomized phase III trial in which KEYTRUDA® administered at a fixed dose was compared with investigator’s choice of cytotoxic chemotherapy, as first line therapy, for patients with advanced NSCLC, with tumor PD-L1 expression of 50% or greater. Three hundred and five (N=305) treatment naïve patients with advanced NSCLC and PD-L1 expression on at least 50% of tumor cells, were randomly assigned in a 1:1 ratio to receive either KEYTRUDA® (N=154) or chemotherapy (N=151). Enrolled patients had no sensitizing EGFR mutations or ALK translocations. Treatment consisted of KEYTRUDA® administered at a fixed dose of 200 mg IV every 3 weeks for 35 cycles or the investigator’s choice of platinum-based chemotherapy for 4-6 cycles. Pemetrexed (ALIMTA®) based therapy was permitted only for patients who had non-squamous tumors and these patients could receive ALIMTA® maintenance therapy after the completion of combination chemotherapy. Patients in the chemotherapy group who experienced disease progression were allowed to cross over to the KEYTRUDA® group. The primary end point was Progression Free Survival (PFS) and secondary end points included Overall Survival (OS), Objective Response Rate (ORR) and safety.
It was previously reported that at a median follow up of 11.2 months, the median PFS was 10.3 months in the KEYTRUDA® group versus 6 months in the chemotherapy group (HR=0.50; P<0.001). However, median OS had not been reached in the KEYTRUDA® group at the time of that analysis. This publication is an updated analysis of the KEYNOTE-024 study, after a median follow-up of 25.2 months. Eighty two patients (N=82) assigned to chemotherapy, met criteria to cross over to the KEYTRUDA® group, upon progression. The median OS was 30 months in the KEYTRUDA® group and 14.2 months in the chemotherapy group (HR=0.63). Further, more patients in the KEYTRUDA® group achieved 12-month OS (70.3% vs. 54.8%) and an ORR response (45.5% vs. 29.8%), compared to the chemotherapy group. The ORR among patients who crossed over to KEYTRUDA®, was 20.7%. The median Duration of Response has not yet been reached for patients assigned to KEYTRUDA® and also for those who crossed over to KEYTRUDA®. For those assigned chemotherapy, the median Duration of Response was 7.1 months. Patients in the KEYTRUDA® group had lower rates of grade 3 to 5 adverse events compared to those in the chemotherapy group (31.2% vs 53.3%), as well as a lower rate of any-grade adverse events (76.6% vs 90%).
It was concluded that first-line treatment with KEYTRUDA® resulted in a significantly longer median OS with lower rates of Adverse Events, when compared to chemotherapy, among patients with metastatic NSCLC and high PD-L1 expression. Brahmer JR, Rodriguez-Abreu D, Robinson A, et al. Updated analysis of KEYNOTE-024: pembrolizumab vs platinum-based chemotherapy for advanced NSCLC with PD-L1 TPS>50%. Presented at: International Association for the Study of Lung Cancer 18th World Conference on Lung Cancer; Yokohama, Japan: October 15-18, 2017. Abstract OA 17.06.
Consolidation with IMFINZI® after Chemoradiotherapy Improves Outcomes in Patients with Unresectable Stage III Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Approximately one third of all patients with NSCLC have stage III, locally advanced disease at the time of initial presentation. Worldwide, about 500,000 patients are diagnosed with unresectable, stage III NSCLC, each year. These patients include those with locally advanced primary tumors with tumor invading the vital mediastinal organs, as well as those with involvement of locoregional mediastinal lymph nodes. These patients are often treated with platinum-based doublet chemotherapy with concurrent radiation and have a median Progression Free Survival (PFS) of approximately 8 months and 5 year survival of only 15%. There is hence a significant unmet need for this patient group, with no major treatment advances thus far.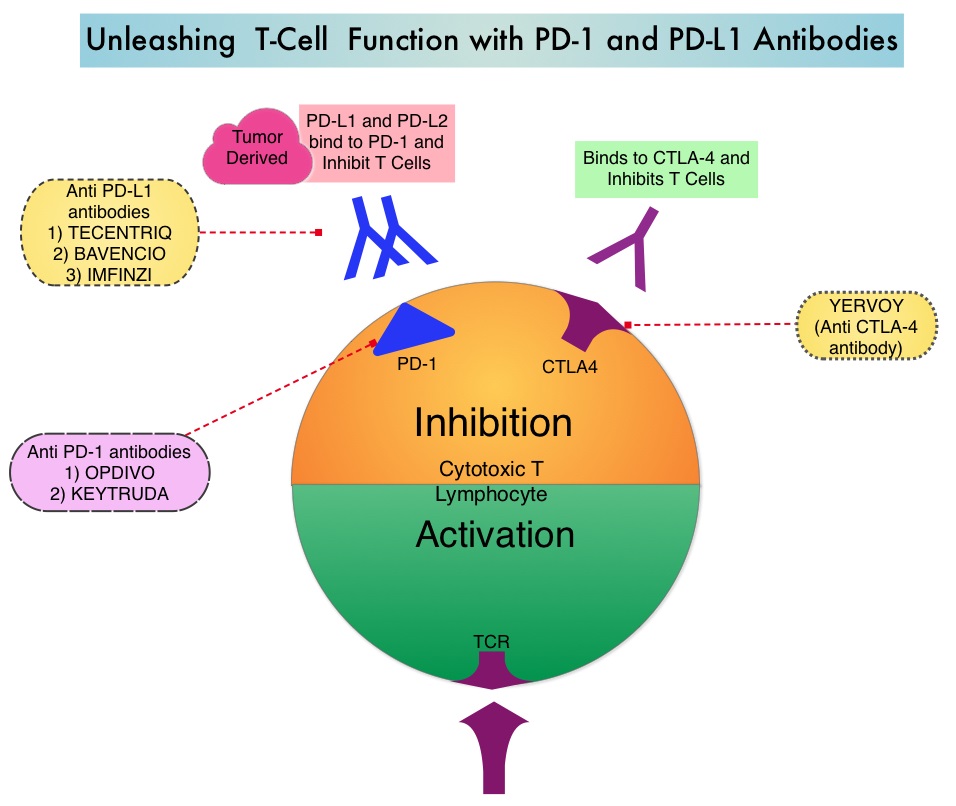
Preclinical evidence had suggested that chemotherapy and radiotherapy may upregulate PD-L1 expression in tumor cells. IMFINZI® (Durvalumab) is a selective, high-affinity, human IgG1 monoclonal antibody, that blocks the binding of Programmed Death Ligand 1 (PD-L1) to Programmed Death 1 (PD-1) and CD80, thereby unleashing the T cells to recognize and kill tumor cells. IMFINZI® showed encouraging antitumor activity in an early phase clinical study involving multiple advanced solid tumors, including stage IIIB or IV NSCLC. IMFINZI® was recently approved by the FDA for the treatment of patients with locally advanced or metastatic urothelial carcinoma, who had received prior platinum-based chemotherapy.
The authors in this publication evaluated the role of immune checkpoint blockade in locally advanced, unresectable, stage III NSCLC. PACIFIC trial is a randomized, double-blind, international, phase III study in which IMFINZI® as consolidation therapy was compared with placebo, in patients with stage III, locally advanced, unresectable NSCLC, that had not progressed following platinum-based chemoradiotherapy. Eligible patients received two or more cycles of platinum-based doublet chemotherapy concurrently with definitive radiation therapy (54-66 Gy). Following completion of concurrent chemoradiation treatment, 713 patients were randomized, of whom 709 patients in a 2:1 ratio received consolidation treatment, within 6 weeks after completion of chemoradiation with IMFINZI® 10 mg/kg every 2 weeks (N=473) or placebo (N=236), for up to 12 months. The median age was 64 years, and the majority of patients were men (70%) and 46% had a squamous histology. The coprimary end points were Progression Free Survival (PFS) and Overall Survival (OS). Secondary end points included 12-month and 18-month PFS rates, Objective Response Rate (ORR), Duration of Response, time to death or distant metastasis, and safety. The authors reported the results of a preplanned interim analysis after a median follow up of 14.5 months.
The median PFS from randomization to consolidation treatment was 16.8 months with IMFINZI® versus 5.6 months with placebo (HR=0.52; P<0.001). This meant a 48% decrease in the probability of disease progression with IMFINZI® and this improvement was consistent across all patient subgroups that were analyzed. The 12-month PFS was 55.9% vs 35.3%, and the 18-month PFS rate was 44.2% vs 27.0%, in favor of IMFINZI®. The ORR was higher with IMFINZI® compared to placebo (28.4% vs 16.0%; P<0.001), and the median Duration of Response was longer as well, with 73% of the patients in the IMFINZI® group having an ongoing response at 18 months versus 47% of the patients in the placebo group. Patients in the IMFINZI® group also had a lower incidence of new brain metastases. The median time to death or distant metastasis was longer with IMFINZI® compared with placebo (23.2 months vs 14.6 months; P<0.001). Adverse events of any grade occurred in 68% of patients in the IMFINZI® group compared to 53% in the placebo group and majority of the toxicities were grade 1 or 2, and grade 3 or higher toxicities were infrequent (less than10%), in both treatment groups. Treatment had to be discontinued due to pneumonitis in 6.3% of patients on IMFINZI® and 4.3% on placebo.
It was concluded that IMFINZI® significantly prolonged PFS in all prespecified groups of patients with locally advanced stage III NSCLC, and toxicity profile was acceptable. Biomarkers, such as mutational load or immunosignature, may be of value, as PD-L1 expression had little or no impact on outcomes. The National Comprehensive Cancer Network (NCCN) Guidelines have been updated to include one year of consolidation therapy with IMFINZI®, after curative-intent chemoradiation, for inoperable stage III lung cancer. Durvalumab after Chemoradiotherapy in Stage III Non-Small Cell Lung Cancer. Antonia SJ, Villegas A, Daniel D, et al. for the PACIFIC Investigators. N Engl J Med 2017; 377:1919-1929
TAGRISSO® Superior to First Generation EGFR TKIs in Advanced Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Approximately 10% to 15% of Caucasian patients and 35-50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R point mutations in Exon 21. EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60% to 70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9 to 14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50% - 60% of patients.
TAGRISSO® (Osimertinib), is a third-generation Epidermal Growth Factor Receptor (EGFR) TKI presently approved by the FDA, for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, who had progressed on prior systemic therapy, including an EGFR-TKI. Previously published studies suggested that TAGRISSO® may also be effective as initial therapy for EGFR mutation-positive advanced NSCLC.
FLAURA is a randomized, double blind, phase III clinical trial, conducted to compare the efficacy and safety of first line TAGRISSO® to TARCEVA® or IRESSA® (which are considered Standard of Care as first line therapy), in NSCLC patients with activating mutations EGFR Exon 19 deletions or L858R substitution mutation on Exon 21. This study randomized 556 advanced NSCLC treatment naïve patients, with EGFR Exon 19 or 21 mutations in a 1:1 ratio, to TAGRISSO® 80 mg orally once daily (N=279) or Standard of Care EGFR-TKI, IRESSA® 250 mg or TARCEVA® 150 mg, orally once daily (N=277). Patients were stratified by mutation status (Exon 19 vs 21 mutations) and race (Asian vs non-Asian). Patients with CNS metastases who were neurologically stable, were allowed in this study. The Primary endpoint was Progression Free Survival (PFS).
The median PFS was 18.9 months with TAGRISSO® compared to 10.2 months for the standard therapy (HR=0.46; P<0.0001), suggesting a 54% reduction in the risk of disease progression, compared with Standard of Care. TAGRISSO® extended the median Time To Progression by about 9 months. This PFS benefit was consistent across all subgroups of patients, including those with and without CNS metastases at study entry. The Objective Response Rate (ORR) with TAGRISSO® was 80% compared with 76% for TARCEVA® and IRESSA®. The median Duration of Response with TAGRISSO® was 17.2 versus 8.5 months in the comparator arm. The median Overall Survival was not reached. Grade 3 and 4 toxicities were lower for TAGRISSO® (34%) compared with 45% for TARCEVA® and IRESSA®. Toxicities led to treatment discontinuation for 13% and 18% of patients in the TAGRISSO® and comparator groups, respectively.
It was concluded that TAGRISSO® demonstrated superior efficacy and tolerability compared to the Standard of Care, as first-line therapy in patients with advanced EGFR mutation positive NSCLC. Studies are underway, assessing treatments, following resistance to TAGRISSO®. Osimertinib vs standard of care (SoC) EGFR-TKI as first-line therapy in patients (pts) with EGFRm advanced NSCLC: FLAURA. Ramalingam S, Reungwetwattana T, Chewaskulyong B, et al. Presented at: 2017 ESMO Congress; Madrid, Spain; September 9-12, 2017. Abstract LBA2_PR.
ASCO Clinical Practice Guideline Update for Stage IV Non Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. ASCO published the last clinical practice guideline update on systemic therapy for patients with Stage IV Non Small Cell Lung Cancer (NSCLC), in 2015. With the many advances in the management of these patients and availability of new practice changing evidence since the last publication, the latest ASCO guideline has been revised. The ASCO NSCLC Expert Panel updated their recommendations based on a systematic review of 14 randomized controlled trials from February 2014 to December 2016 and six nonrandomized studies on systemic therapy. This guideline is applicable to patients who had received molecular testing for EGFR/ALK/ROS1, as well as Programmed Death Ligand 1 (PD-L1), and clinicians know the test results.
Guideline Question: What systemic therapy treatment options should be offered to patients with Stage IV NSCLC, depending on the subtype of the patient’s cancer?
Target Population: Patients with Stage IV NSCLC.
Target Audience: Oncology care providers (including primary care physicians, specialists, nurses, social workers, and any other relevant member of a comprehensive multidisciplinary cancer care team), patients, and their caregivers.
Key Points:
1) There is no cure for patients with Stage IV NSCLC.
2) Decisions regarding chemotherapy should not be made based on age alone.
Recommendations: First Line Treatment for Patients
Patients with Non-Squamous Cell Carcinoma without a tumor EGFR-sensitizing mutation or ALK or ROS1 gene rearrangement and with a Performance Status (PS) of 0 or 1 (and appropriate PS of 2):
1) With high PD-L1 expression (Tumor Proportion Score [TPS] 50% or more) and no contraindications, single-agent Pembrolizumab is recommended.
2) With low PD-L1 expression (TPS less than 50%), a variety of combination cytotoxic chemotherapies (with or without Bevacizumab, if patients are receiving Carboplatin and Paclitaxel) are recommended.
3) There is insufficient evidence to recommend Bevacizumab in combination with Pemetrexed plus Carboplatin.
4) Other checkpoint inhibitors, combination checkpoint inhibitors, or immune checkpoint therapy with chemotherapy are not recommended.
5) With PS of 2, combination or single agent therapy or palliative care alone may be used.
Patients with Squamous Cell Carcinoma without a tumor EGFR-sensitizing mutation or ALK or ROS1 gene rearrangement and with a PS of 0 or 1 (and appropriate PS of 2):
1) With high PD-L1 expression (TPS 50% or more) and no contraindications, single agent Pembrolizumab is recommended.
2) With low PD-L1 expression (TPS less than 50%), a variety of combination cytotoxic chemotherapies are recommended.
3) Other checkpoint inhibitors, combination checkpoint inhibitors, or immune checkpoint therapy with chemotherapy are not recommended.
4) With PS of 2, combination or single agent therapy or palliative care alone may be used.
5) With Squamous NSCLC treated with Cisplatin and Gemcitabine, the Panel neither recommends for nor recommends against the addition of Necitumumab to chemotherapy.
With sensitizing EGFR mutations, Afatinib, Erlotinib, or Gefitinib is recommended.
With ALK gene rearrangements, Crizotinib is recommended.
With ROS1 rearrangement, Crizotinib is recommended.
Recommendations: Second Line Treatment for Patients
Without a tumor EGFR-sensitizing mutation or ALK or ROS1 gene rearrangement and with PS of 0 or 1 (and appropriate PS of 2):
1) In patients with high PD-L1 expression (TPS 1% or more), no contraindications, who received first line chemotherapy and have not received prior immune therapy, single agent Nivolumab, Pembrolizumab, or Atezolizumab is recommended.
2) In patients with negative or unknown tumor PD-L1 expression (TPS less than 1%), no contraindications and who received first line chemotherapy, single agent Nivolumab, or Atezolizumab, or a variety of combination cytotoxic chemotherapies are recommended.
3) Other checkpoint inhibitors, combination checkpoint inhibitors, and immune checkpoint therapy with chemotherapy are not recommended.
4) In patients who received an immune checkpoint inhibitor as first line therapy, a variety of combination cytotoxic chemotherapies are recommended.
5) In patients with contraindications to immune checkpoint inhibitor therapy after first line chemotherapy, Docetaxel is recommended.
6) In patients with non-Squamous Cell Carcinoma who have not previously received Pemetrexed, Pemetrexed is recommended.
With sensitizing EGFR mutations:
1) In patients with disease progression after first line therapy with an EGFR Tyrosine Kinase Inhibitor (TKI) and the presence of the T790M resistance mutation, Osimertinib is recommended. If T790M mutation is not present, a platinum doublet is recommended.
2) In patients who received an EGFR-TKI in the first-line setting, had an initial response, and subsequently experienced slow or minimal disease progression at isolated sites, EGFR-TKI with local therapy to the isolated sites is an option.
With ROS1 rearrangement:
1) In patients who have not received prior Crizotinib, Crizotinib is recommended.
2) In patients who have received prior Crizotinib, platinum-based therapy in the second line with or without Bevacizumab is recommended.
With BRAF mutations:
1) In patients without prior immune checkpoint therapy and high PD-L1 expression (TPS more than 1%), single agent Atezolizumab, Nivolumab, or Pembrolizumab is recommended.
2) In patients who have received prior immune checkpoint therapy, Dabrafenib alone or in combination with Trametinib in third line, is an option.
Recommendations: Third Line Treatment for Patients
1) In patients without a tumor EGFR-sensitizing mutation or ALK or ROS1 gene rearrangement and with non-Squamous Cell Carcinoma and PS of 0 or 1 (and appropriate PS of 2), who received chemotherapy with or without Bevacizumab and immune checkpoint therapy, single agent Pemetrexed or Docetaxel are options.
2) In patients with tumor EGFR-sensitizing mutation(s) who have received at least one first-line EGFR-TKI and prior platinum-based chemotherapy, there are insufficient data to recommend immunotherapy in preference to chemotherapy.
Recommendations: Fourth Line Treatment for Patients
Patients and clinicians should consider and discuss experimental treatment, clinical trials, and continued best supportive (palliative) care.
Systemic Therapy for Stage IV Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. Hanna N, Johnson D, Temin S, et al. J Clin Oncol 2017;35:3484-3515
FDA Approves TAFINLAR® and MEKINIST® Combo for BRAF V600E-Mutant Non Small Cell Lung Cancer
SUMMARY: The FDA on June 22, 2017 granted regular approvals to TAFINLAR® ((Dabrafenib) and MEKINIST® (Trametinib) administered in combination, for patients with metastatic Non Small Cell Lung Cancer (NSCLC) with BRAF V600E mutation, as detected by an FDA-approved test. These are the first FDA approvals specifically for treatment of patients with BRAF V600E mutation-positive metastatic NSCLC. The FDA also approved the Oncomine® Dx Target Test (Thermo Fisher Scientific), a Next Generation Sequencing (NGS) test to detect multiple gene mutations for lung cancer in a single test from a single tissue specimen. This test detects the presence of BRAF, ROS1, and EGFR gene mutations or alterations in tumor tissue of patients with NSCLC. This test can be used to select patients with NSCLC with the BRAF V600E mutation for treatment with the combination of TAFINLAR® and MEKINIST®. This is the first NGS oncology panel test approved by the FDA for multiple companion diagnostic indications. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States.
The approval of the combination of MEKINIST® (Trametinib) and TAFINLAR® (Dabrafenib), to treat patients with advanced NSCLC, was based on the understanding of the biological pathways of this malignancy. The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been demonstrated in 6%-8% of all malignancies and BRAF V600E mutation occurs in 1-2% of lung adenocarcinomas and acts as an oncogenic driver.
BRF113928 is an open-label, multicohort, multicentre, non-randomized, phase II study which sequentially enrolled patients with BRAF V600E mutation-positive metastatic NSCLC, across 3 cohorts. The first 2 cohorts included previously treated patients. The median age was 64 years, 98% had adenocarcinoma, and majority of patients (72%) were former or current smokers. In the first cohort, 84 patients received single agent TAFINLAR® following one or more prior platinum-based chemotherapy. In the second cohort, 57 patients received a combination of TAFINLAR® 150 mg orally twice daily and MEKINIST® 2 mg orally once daily. Patients in this second cohort had received at least 1 prior line of platinum-based chemotherapy regimen with disease progression and 33% had received 2 or more prior chemotherapy regimens.
It was noted that in these previously treated cohorts of patients, the Objective Response Rate (ORR) for the combination treatment based on independent review was 63%, with a median Duration of Response (DoR) of 12.6 months. The ORR for patients who received single agent TAFINLAR® was 27% and the median DoR was 9.9 months. The investigator assessed median Progression Free Survival (PFS) was 10.2 months, and median Overall Survival (OS) was 18.2 months.
The authors in this latest publication reported the the efficacy and safety of TAFINLAR® plus MEKINIST® treatment in previously untreated patients with BRAF V600E-mutant metastatic NSCLC (third cohort). This cohort enrolled 36 patients and patients received TAFINLAR® 150 mg orally twice daily plus MEKINIST® 2 mg orally once daily, until disease progression or unacceptable toxicities. The median follow up was 15.9 months. The Primary endpoint was investigator assessed ORR. Secondary endpoints included Duration of Response (DoR), PFS, OS, and safety.
The Objective Response Rate in this cohort was 64%, with 6% Complete Response and 58% Partial Response. The Disease Control Rate was 75%. The median Duration of Response was 10.4 months, median PFS was 10.9 months and median OS was 24.6 months. The most common grade 3 or 4 adverse events were pyrexia, liver function abnormalities, hypertension and vomiting.
The authors concluded that TAFINLAR® plus MEKINIST® demonstrated substantial antitumor activity and durable responses in patients with treatment-naive BRAF V600E-mutant NSCLC. This study confirmed that there is a fourth actionable biomarker, BRAF V600E, in addition to EGFR, ALK and ROS-1, for patients with Non Small Cell Lung Cancer. Dabrafenib plus trametinib in patients with previously untreated BRAF V600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Planchard D, Smit EF, Groen HJ, et al. The Lancet Oncology 2017;18:1307-1316
ASTRO Guideline for Stereotactic Body Radiation Therapy in Early Stage Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Approximately 15% of patients present with early stage (T1-2 N0) disease, and these numbers are likely to increase with the implementation of Lung Cancer screening programs. Patients with early stage disease unless medically unfit, undergo surgical resection with a curative intent. Those who are not surgical candidates, are often treated with conventional Radiation Therapy, which can result in high rates of local failure and treatment-related toxicities.
Stereotactic Body Radiation Therapy (SBRT) is a non-surgical procedure that allows delivery of significantly higher doses of precisely focused radiation to the tumor, compared to conventional Radiation Therapy, with less collateral damage to the surrounding normal tissue. The technologies used for SBRT include GAMMA KNIFE® which uses highly focused gamma rays, Proton Beam therapy which uses ionized hydrogen or Protons, Linear Accelerator (LINAC) and CYBER KNIFE® which use Photons, to target the tumor tissue. Because SBRT is fractionated and delivered over 1-5 days, the short-and long-term side effects of radiation therapy are decreased and may allow higher total dosage to be given.
This guideline is based on systematic review of literature which included 172 articles, from January 1995 and August, 2016. This literature search evaluated adults with T1-2, N0, Non Small Cell Lung Cancer (NSCLC) receiving primary or salvage SBRT. Developed by the American Society for Radiation Oncology, this guideline is also endorsed by the European Society for Radiotherapy & Oncology, the Royal Australian and New Zealand College of Radiologists, and the International Association for the Study of Lung Cancer.
KEY QUESTIONS (KQ)
KQ 1: When is SBRT appropriate for patients with T1-2, N0, NSCLC who are medically operable?
Statement KQ 1A: Any patient with operable Stage I NSCLC being considered for SBRT should be evaluated by a thoracic surgeon, preferably in a multidisciplinary setting, to reduce specialty bias.
Statement KQ 1B: For patients with “standard operative risk” (ie, with anticipated operative mortality of <1.5%) and stage I NSCLC, SBRT is not recommended as an alternative to surgery outside of a clinical trial. Discussions about SBRT are appropriate, with the disclosure that long-term outcomes with SBRT >3 years are not well established. For this population, lobectomy with systematic mediastinal lymph node evaluation remains the recommended treatment, though a sublobar resection may be considered in select clinical scenarios.
Statement KQ 1C: For patients with “high operative risk” (ie, those who cannot tolerate lobectomy, but are candidates for sublobar resection) stage I NSCLC, discussions about SBRT as a potential alternative to surgery are encouraged. Patients should be informed that while SBRT may have decreased risks from treatment in the short term, the longer term outcomes >3 years are not well-established.
KQ 2: When is SBRT appropriate for medically inoperable patients with T1-2, N0, NSCLC?
For patients with centrally located tumors
Statement KQ 2A: SBRT directed toward centrally located lung tumors (tumor within 2 cm of the proximal tracheobronchial tree) carries unique and significant risks when compared to treatment directed at peripherally located tumors. The use of 3-fraction regimens should be avoided in this setting.
Statement KQ 2B: SBRT directed at central lung tumors should be delivered in 4 or 5 fractions. Adherence to volumetric and maximum dose constraints may optimize the safety profile of this treatment. For central tumors for which SBRT is deemed too high risk, hypofractionated radiation therapy utilizing 6 to 15 fractions can be considered.
For patients with tumors >5 cm in diameter
Statement KQ 2C: SBRT is an appropriate option for tumors >5 cm in diameter with an acceptable therapeutic ratio. Adherence to volumetric and maximum dose constraints may optimize the safety profile of this treatment.
For patients lacking tissue confirmation
Statement KQ 2D: Whenever possible, obtain a biopsy prior to treatment with SBRT to confirm a histologic diagnosis of a malignant lung nodule.
Statement KQ 2E: SBRT can be delivered in patients who refuse a biopsy, have undergone non-diagnostic biopsy, or who are thought to be at prohibitive risk of biopsy. Prior to SBRT in patients lacking tissue confirmation of malignancy, patients are recommended to be discussed in a multidisciplinary manner with a consensus that the lesion is radiographically and clinically consistent with a malignant lung lesion based on tumor, patient, and environmental factors
For patients with synchronous primary or multifocal tumors
Statement KQ 2F: Multiple Primary Lung Cancers (MPLCs) can be difficult to differentiate from intrathoracic metastatic lung cancer and pose unique issues for parenchymal preservation; therefore, it is recommended that they are evaluated by a multidisciplinary team.
Statement KQ 2G: Positron Emission Tomography/Computed Tomography and brain Magnetic Resonance Imaging are recommended in patients suspected of having MPLC to help differentiate from intrathoracic metastatic lung cancer. Invasive mediastinal staging should be addressed on a case-by-case basis.
Statement KQ 2H: SBRT may be considered as a curative treatment option for patients with synchronous MPLC. SBRT for synchronous MPLC has equivalent rates of local control and toxicity, but decreased rates of overall survival compared with those with single tumors.
Statement KQ 2I: SBRT is recommended as a curative treatment option for patients with metachronous MPLC. SBRT for metachronous MPLC has equivalent rates of local control and toxicity and overall survival compared with those with single tumors.
For patients who underwent pneumonectomy and now have a new primary tumor in their remaining lung
Statement KQ 2J: SBRT may be considered a curative treatment option for patients with metachronous MPLC in a postpneumonectomy setting. While SBRT for metachronous MPLC appears to have equivalent rates of local control and acceptable toxicity compared to single tumors, SBRT in the post-pneumonectomy setting might have a higher rate of toxicity than in patients with higher baseline lung capacity.
KQ 3: For medically inoperable early-stage lung cancer patients, how can SBRT techniques be individually tailored to provide an adequate dose for tumor eradication with minimal risk to normal structures in “high-risk” clinical scenarios?
For tumors with intimal proximity/involvement of mediastinal structures (bronchial tree, esophagus, heart, etc.)
Statement KQ 3A: For tumors in close proximity to the proximal bronchial tree, SBRT should be delivered in 4 to 5 fractions. Physicians should endeavor to meet the constraints that have been utilized in prospective studies given the severe toxicities that have been reported.
Statement KQ 3B: For tumors in close proximity to the esophagus, physicians should endeavor to meet the constraints that have been utilized in prospective studies or otherwise reported in the literature given the severe esophageal toxicities that have been reported.
Statement KQ 3C: For tumors in close proximity to the heart and pericardium, SBRT should be delivered in 4 to 5 fractions with low incidence of serious toxicities to the heart, pericardium, and large vessels observed. Adherence to volumetric and maximum dose constraints utilized in prospective trials or reported in the literature may optimize the safety profile of this treatment.
For tumors abutting or invading the chest wall
Statement KQ 3D: SBRT is an appropriate option for treatment and should be offered for T1-2 tumors that abut the chest wall. Grade 1 and 2 chest wall toxicity is a common occurrence post SBRT that usually resolves with conservative management. Patients with peripheral tumors approximating the chest wall should be counseled on the possibility of this common toxicity.
Statement KQ 3E: SBRT may be utilized in patients with cT3 disease due to chest wall invasion without clear evidence of reduced efficacy or increased toxicity compared to tumors abutting the chest wall.
KQ 4: In medically inoperable patients, what is the role of SBRT as salvage therapy for early-stage lung cancer that recurs?
After conventionally fractionated Radiation Therapy
Statement KQ 4A: The use of salvage SBRT after primary conventionally fractionated radiation may be offered to selected patients due to reported favorable local control and survival. These patients should be informed of significant (including fatal) toxicities.
Statement KQ 4B: Patient selection for salvage SBRT after primary conventionally fractionated radiation is a highly individualized process. Radiation oncologists should assess evidence-based patient, tumor, and treatment factors prior to treatment initiation.
After SBRT and sublobar resection
Statement KQ 4C: Patient selection for salvage SBRT after previous SBRT and after prior Sublobar resection is a highly individualized process. Radiation oncologists should assess evidence-based patient, tumor, and treatment factors before treatment initiation.
Stereotactic Body Radiation Therapy for early-stage Non-Small Cell Lung Cancer: Executive Summary of an ASTRO Evidence-Based Guideline. Videtic GM, Donington J, Giuliani M, et al. http://dx.doi.org/10.1016/j.prro.2017.04.014
FDA Approves TAFINLAR® and MEKINIST® Combination for BRAF Positive Non Small Cell Lung Cancer
SUMMARY: The FDA on June 22, 2017, granted regular approvals to TAFINLAR® (Dabrafenib) and MEKINIST® (Trametinib) administered in combination, for patients with metastatic Non Small Cell Lung Cancer (NSCLC), with BRAF V600E mutation, as detected by an FDA-approved test. These are the first FDA approvals specifically for treatment of patients with BRAF V600E mutation-positive metastatic NSCLC.
The FDA also approved the Oncomine® Dx Target Test, a next generation sequencing (NGS) test to detect multiple gene mutations for lung cancer in a single test from a single tissue specimen. This test detects the presence of BRAF, ROS1, and EGFR gene mutations or alterations in tumor tissue of patients with NSCLC. This test can be used to select patients with NSCLC with the BRAF V600E mutation for treatment with the combination of TAFINLAR® and MEKINIST®. This is the first NGS oncology panel test approved by the FDA for multiple companion diagnostic indications.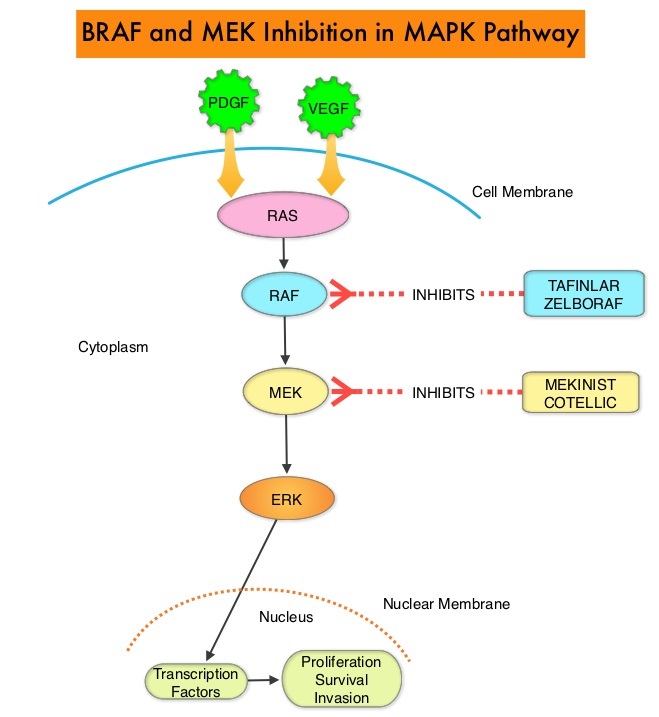
Combining MEKINIST® (Trametinib) with TAFINLAR® (Dabrafenib) to treat patients with NSCLC, was based on the understanding of the biological pathways of this malignancy. The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. This pathway has been implicated in the development of multiple malignancies including NSCLC and Melanoma. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been demonstrated in 6-8% of all malignancies. TAFINLAR® is a selective oral BRAF inhibitor and MEKINIST® is a potent and selective inhibitor of MEK gene, which is downstream from RAF in the MAPK pathway.
The approval of TAFINLAR® and MEKINIST® combination, for patients with metastatic NSCLC was based on an international, multicenter, three-cohort, non-randomized, open-label trial, in patients with locally confirmed BRAF V600E mutation-positive, metastatic NSCLC. In this phase II trial, 93 patients were treated with the combination of TAFINLAR® 150 mg orally twice daily and MEKINIST® 2 mg orally once daily. Of these 93 patients, 36 patients had received no prior systemic therapy for metastatic NSCLC and 57 patients received at least one prior platinum-based chemotherapy regimen and had disease progression. The third cohort in this phase II trial included 78 previously treated patients with BRAF V600E mutation-positive metastatic NSCLC, who received single-agent TAFINLAR®. The primary endpoint was Overall Response Rate (ORR).
It was noted that in the previously treated group, the ORR for the combination treatment based on independent review was 63% with a median Duration of Response of 12.6 months. In the treatment-naive group, the ORR for the combination was 61% and this group had not reached the endpoint for median Duration of Response and therefore was not estimable. However, among those who responded to treatment, 59% of the responders had response durations greater than 6 months. The ORR for patients who received single agent TAFINLAR® was 27% and the median Duration of Response was 9.9 months. The most common Grade 3-4 adverse reactions were pyrexia, fatigue, dyspnea, vomiting, rash, hemorrhage, and diarrhea.
It was concluded that TAFINLAR® plus MEKINIST® combination represents a new targeted therapy for patients with BRAF V600E mutation¬-positive metastatic NSCLC, who tend to respond less favorably to standard chemotherapy. This approval marks the fourth actionable genomic biomarker in metastatic NSCLC along with EGFR, ALK and ROS-1. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Planchard D, Besse B, Groen HJ et al. Lancet Oncol. 2016 Jul;17(7):984-93. doi: 10.1016/S1470-2045(16)30146-2. Epub 2016 Jun 6.
Late Breaking Abstract – ASCO 2017: Dacomitinib Superior to IRESSA® in EGFR Mutant Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Approximately 10% to 15% of Caucasian patients and 50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R point mutations in Exon 21. EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60% to 70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations.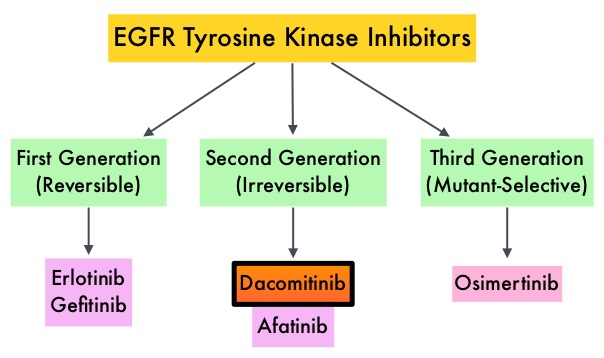
Dacomitinib is a potent, irreversible, second-generation EGFR Tyrosine Kinase Inhibitor and inhibits three members of the ErbB protein family, including EGFR/HER1, HER2 and HER4. Based on the encouraging clinical activity of Dacomitinib in treatment naïve patients with advanced NSCLC, harboring activating EGFR mutations, in a phase II study (The Lancet Oncology 2014;15:1433-1441), the authors conducted a randomized phase III trial, comparing Dacomitinib with IRESSA®, as first line therapy in this patient population . This study (ARCHER 1050) randomized 452 patients in a 1:1 ratio to either receive Dacomitinib 45 mg PO daily (N=227) or IRESSA® 250 mg PO daily (N=225). Eligible patients had newly diagnosed stage IIIB/IV or recurrent NSCLC, harboring an activating EGFR mutation (Exon 19 deletions or L858R point mutations in Exon 21, with or without Exon 20 T790M mutations). Treatments groups were well balanced and patients were stratified by race and EGFR mutation subtype. The Primary endpoint was Progression Free Survival (PFS) and Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR) and Duration of Response (DoR).
The median PFS for patients who received Dacomitinib was 14.7 months compared with 9.2 months for the group who received IRESSA® (HR=0.59; P<0.0001). This meant a 41% reduction in the risk of cancer progression or death with Dacomitinib compared with IRESSA®. The median Duration of Response was 14.8 months with Dacomitinib versus 8.3 months with IRESSA® (HR= 0.40; P<0.0001). As expected, patients in the Dacomitinib group experienced more side effects such as skin rash and diarrhea and this has been attributed to the stronger suppression of the EGFRs in the normal healthy tissues.
The authors concluded that ARCHER 1050 is the first phase III trial comparing EGFR TKIs head-to-head, and this study demonstrated clinically meaningful superiority of Dacomitinib, when compared to IRESSA®, in treatment naïve NSCLC patients, with activating EGFR mutations. Further, the PFS achieved with Dacomitinib in this study is among the highest observed, when compared with other EGFR Tyrosine Kinase Inhibitors, for this cancer type. Dacomitinib versus gefitinib for the first-line treatment of advanced EGFR mutation positive non-small cell lung cancer (ARCHER 1050): A randomized, open-label phase III trial. Mok T, Cheng Y, Zhou X, et al. J Clin Oncol 35, 2017 (suppl; abstr LBA9007)
Late Breaking Abstract - ASCO 2017: ALECENSA® Superior to XALKORI® in Untreated ALK-Positive Non Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous cell carcinomas, 40% are Adenocarcinomas and 10% are Large cell carcinomas. The discovery of rearrangements of the Anaplastic Lymphoma Kinase (ALK) gene in some patients with advanced NSCLC and adenocarcinoma histology, led to the development of agents such as XALKORI® (Crizotinib), ZYKADIA® (Ceritinib), ALECENSA® (Alectinib) and now ALUNBRIG® (Brigatinib), with promising results. It has become clear that appropriate, molecularly targeted therapy for tumors with a molecular abnormality, results in the best outcomes. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8% as well as other mutations in BRAF, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive and the presence of two simultaneous mutations, are rare.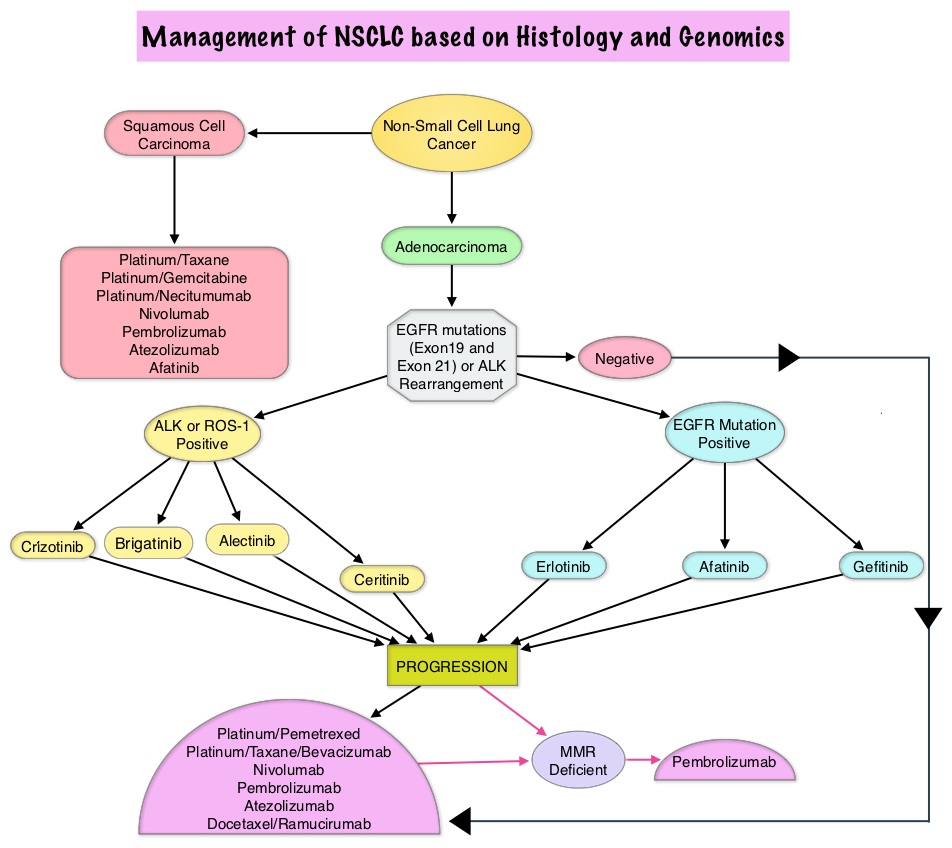
Patients with advanced NSCLC harboring ALK gene rearrangements often receive XALKORI® as first line therapy and can expect a median Progression Free Survival of approximately 11 months. These patients however are at a high lifetime risk of CNS metastases. ALECENSA® (Alectinib) is a potent ALK Tyrosine Kinase Inhibitor, and is effective against several ALK mutations that confer resistance to XALKORI® Further, unlike XALKORI®, ALECENSA® can cross the blood-brain barrier and is not a substrate of P-glycoprotein, a key efflux transporter located at the blood-brain barrier.
The ALEX trial is an international, randomized, open-label, phase III study which compared ALECENSA® with XALKORI®, in patients with previously untreated, advanced ALK-positive NSCLC, including those with asymptomatic CNS disease. In this trial, 303 previously untreated patients were randomly assigned in a 1:1 ratio to receive ALECENSA® at 600 mg twice daily (N=152) or XALKORI® at 250 mg PO twice daily (N=151). Treatment was continued until disease progression or unacceptable toxicities. Patients with isolated asymptomatic CNS progression could receive a local therapy at the investigator’s discretion, followed by continued trial treatment until systemic disease progression. Patients were stratified and the primary end point was Investigator-assessed Progression Free Survival. Secondary end points were Independent Review Committee (IRC)–assessed Progression Free Survival, time to CNS progression, Objective Response Rate, and Overall Survival.
At the time of primary analysis, ALECENSA® was significantly superior to XALKORI®, reducing the risk of progression/death by 53% (HR= 0.47; P<0.0001). The median PFS for ALECENSA® was Not Reached versus 11.1 months for XALKORI®. The median Progression Free Survival assessed by the IRC was 25.7 months for ALECENSA® vs 10.4 months for XALKORI® (HR=0.50, P< 0.001). The magnitude of the benefit with ALECENSA® was generally consistent across all the subgroups although this benefit was lower in the subgroups of active smokers and patients with poor Performance Status. Objective Response Rate was 82.9% in the ALECENSA® group versus 75.5% in the XALKORI® group (P=0.09). The rate of CNS progression was 12% in the ALECENSA® group compared with 45% in the XALKORI® group (HR=0.16; P<0.001). Among patients with measurable or non-measurable CNS lesions at baseline, a CNS response occurred in 59% of the patients in the ALECENSA® group versus 26% in the XALKORI® group. Further, 45% of the patients in the ALECENSA® group had a complete CNS response, as compared with 9% in the XALKORI® group. Grade 3-5 adverse events were less frequent with ALECENSA® (41%) versus 50% with XALKORI®.
It was concluded that ALECENSA® showed superior efficacy and lower toxicity compared with XALKORI®, and should be a new standard of care for treatment-naïve patients with ALK-positive NSCLC. Alectinib versus crizotinib in treatment-naive advanced ALK-positive non–small cell lung cancer (NSCLC): primary results of the global phase III ALEX study. Shaw AT, Peters S, Mok T, et al. J Clin Oncol. 2017;35 (suppl; abstr LBA9008).
FDA Approves KEYTRUDA® in Combination with Chemotherapy as First-Line Treatment for Metastatic NSCLC
SUMMARY: The FDA on May 10, 2017 granted accelerated approval to KEYTRUDA® (Pembrolizumab) in combination with ALIMTA® (Pemetrexed) and Carboplatin, for the treatment of patients with previously untreated metastatic non-squamous Non Small Cell Lung Cancer (NSCLC). Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Non Small Cell Lung Cancer accounts for approximately 85% of all lung cancers. The FDA in October 2016, approved KEYTRUDA® for the treatment of patients with metastatic NSCLC, whose tumors have high PD-L1 expression (Tumor Proportion Score greater than or equal to 50%), as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and no prior systemic chemotherapy treatment for metastatic NSCLC.
KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as  membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.
membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.
This recent additional FDA approval for KEYTRUDA®, was based on data from the KEYNOTE-021 trial, which is a randomized, open-label, phase 2 cohort of a multicohort study. Chemotherapy-naive, Stage IIIB or IV, non-squamous NSCLC patients (N=123),with no targetable EGFR or ALK genetic aberrations, were randomly assigned in a 1:1 ratio to receive to KEYTRUDA® 200 mg, IV along with Carboplatin AUC 5, IV and ALIMTA® 500 mg/m2 IV every 3 weeks, for 4 cycles, followed by KEYTRUDA® for 24 months and indefinite ALIMTA® maintenance therapy (N=60) or to 4 cycles of Carboplatin and ALIMTA® alone followed by indefinite ALIMTA® maintenance therapy (N=63). The median age was 63 years and patients were stratified by PD-L1 Tumor Proportion Score (<1% versus 1% or more). Approximately 25% of patients in the KEYTRUDA® group were never smokers versus 14% in the control group. The Primary endpoint was Objective Response Rate assessed by an Independent Central Review.
The addition of KEYTRUDA® to Carboplatin and ALIMTA® resulted in an Objective Response Rate (ORR) that was nearly double the ORR of Carboplatin and ALIMTA® alone. (55% versus 29% respectively, P=0.0032). All responses were partial responses. Among patients who received the triplet regimen, 93% had a response duration of 6 months or longer compared to 81% who received Carboplatin and ALIMTA® alone. Additionally, there was a significant improvement in the Progression Free Survival (PFS), with a median PFS of 13.0 months for the KEYTRUDA® combination group compared with 8.9 months for the Carboplatin and ALIMTA® alone group (HR=0.53, P=0.02). Subset analysis revealed that in the subgroup with a PD-L1 Tumor Proportion Score (TPS) of less than 1%, the ORR was 57% with the KEYTRUDA® combination versus 13% for the Carboplatin and ALIMTA® alone group. In the TPS of 1% or more subgroup, the ORR was 54% in the KEYTRUDA® plus Carboplatin and ALIMTA® group and 38% in the Carboplatin and ALIMTA® alone group. There was a higher incidence of adverse events with the addition of KEYTRUDA® to Carboplatin and ALIMTA® with the most common toxicities being fatigue, nausea and constipation, rash and dyspnea.
The authors concluded that a combination of KEYTRUDA®, Carboplatin and ALIMTA® is an effective and tolerable first-line treatment option for patients with advanced non-squamous NSCLC, irrespective of PD-L1 expression. These findings are being explored in an ongoing randomized, Phase III study. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Langer CJ, Gadgeel SM, Borghaei H, et al. Lancet Oncol. 2016;17:1497-1508.
FDA Approves ALUNBRIG® for ALK Positive Non Small Cell Lung Cancer
SUMMARY: The FDA on April 28, 2017 granted accelerated approval to ALUNBRIG® (Brigatinib),for the treatment of patients with metastatic Anaplastic Lymphoma Kinase (ALK)-positive Non Small Cell Lung Cancer (NSCLC), who have progressed on or are intolerant to XALKORI® (Crizotinib). Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 222,500 new cases of lung cancer will be diagnosed in the United States in 2017 and over 155,870 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 25% are Squamous Cell Carcinomas, 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. The discovery of rearrangements of the Anaplastic Lymphoma Kinase (ALK) gene in some patients with advanced NSCLC and adenocarcinoma histology, led to the development of agents such as XALKORI® (Crizotinib), ZYKADIA® (Ceritinib), ALECENSA® (Alectinib) and now ALUNBRIG® (Brigatinib), with promising results. It has become clear that appropriate, molecularly targeted therapy for tumors with a molecular abnormality, results in the best outcomes. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8% as well as other mutations in BRAF, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive, and the presence of two simultaneous mutations are rare.
The approval was based on findings from the ALTA trial, which is a pivotal, open-label multicenter study in which 222 patients were randomized 1:1 ratio to receive ALUNBRIG® 90 mg orally once daily (N=112) or 180 mg once daily following a 7-day lead-in at 90 mg orally once daily (N=110). Two dose regimens were evaluated in this study, as clinical responses and Adverse Events varied with starting dose of ALUNBRIG®, in a previous Phase I/II study. Eligible patients had locally advanced or metastatic ALK-positive NSCLC, who had progressed on XALKORI®. The median age of patients across the study was 54 years and 67% of the patients had brain metastases. Patients were stratified by presence of brain metastases at baseline and best response to prior treatment with XALKORI®. The Primary endpoint was Objective Response Rate (ORR) and Secondary endpoints included Progression Free Survival (PFS) and CNS response.

The ORR was 48% in the 90 mg dose group and 53% in the 180 mg dose group. After a median duration of follow up of 8 months, median duration of response was 13.8 months in both treatment groups. In patients with measurable brain metastases at baseline, intracranial ORR was 42% in the 90 mg group and 67% in the 180 mg group. Among patients who exhibited an intracranial response, 78% of patients in the 90 mg group and 68% of patients in the 180 mg group maintained an intracranial response for at least 4 months. The median PFS was 8.8 months in the 90 mg dose group, and 11.1 months in the 180mg dose group. The most common adverse reactions, were nausea, diarrhea, fatigue, cough, and headache. The most common serious adverse reactions were pneumonia and pneumonitis.
The authors concluded that treatment with ALUNBRIG® resulted in significant response rates and improved Progression Free Survival, with acceptable toxicity profile. The recommended dosing regimen of ALUNBRIG® is 90 mg orally once daily for the first 7 days and if tolerated, the dose is increased to 180 mg orally once daily. Kim D-W, Tiseo M, Ahn M-J, et al. Brigatinib (BRG) in patients (pts) with crizotinib (CRZ)-refractory ALK+ non-small cell lung cancer (NSCLC): First report of efficacy and safety from a pivotal randomized phase (ph) 2 trial (ALTA). J Clin Oncol. 2016;34 (suppl; abstr 9007).
Patients with Lung Cancer and Liver Metastases May Not Benefit from OPDIVO®
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Non Small Cell Lung Cancer accounts for approximately 85% of all lung cancers. The treatment paradigm for malignancies has been rapidly evolving, with a better understanding of the Immune checkpoints or gate keepers. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, Immune checkpoints or gate keepers inhibit an intense immune response by switching off the T cells of the immune system. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are now available that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), as well as Programmed cell Death Ligands (PD-L1), that are expressed by cells in the tumor micro environment. By targeting the Immune check point proteins or their ligands, T cells are unleashed, resulting in T cell proliferation, activation and a therapeutic response.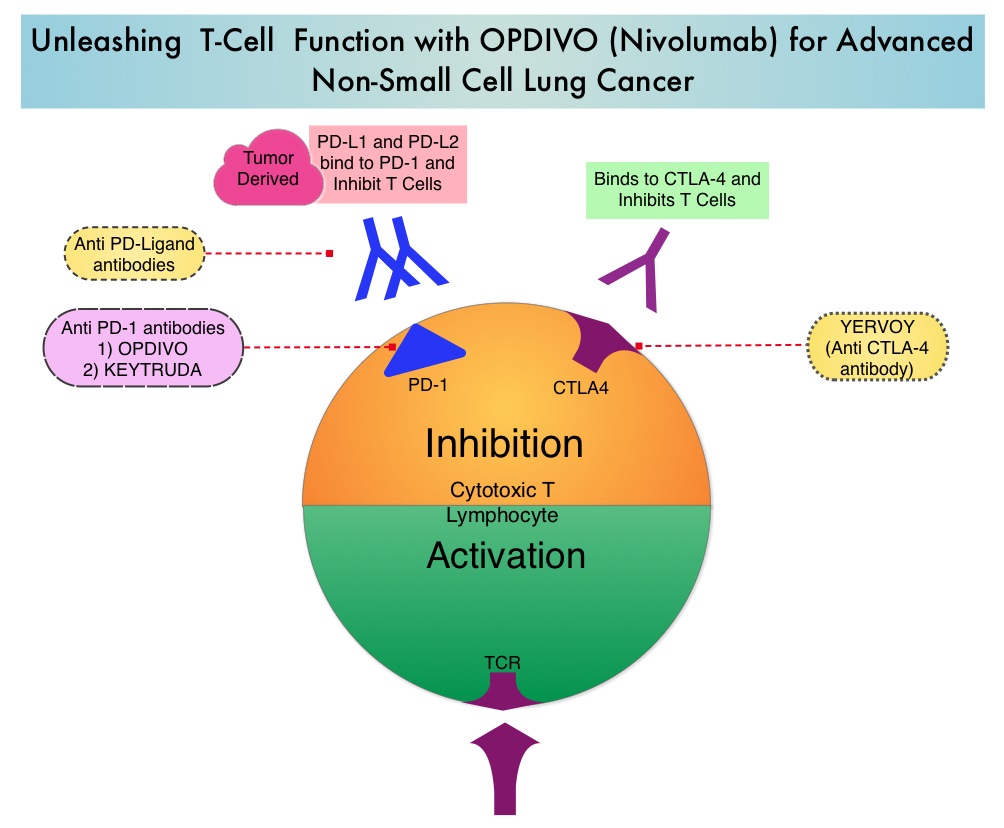
OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. Even though immune checkpoint inhibition has taken center stage in the management of advanced lung cancer as well as a number of other malignancies, the efficacy of these agents appears to vary based on the site of metastatic disease. Previously published studies have shown that in patients with advanced NSCLC, adrenal lesions and lymph nodes were more responsive when treated with OPDIVO®, followed by lung lesions. Liver lesions were less responsive to OPDIVO®. (Nishino M, Ramaiya NH, Chambers ES, et al. Immune-related response assessment during PD-1 inhibitor therapy in advanced non-small-cell lung cancer patients. Journal for Immunotherapy of Cancer. 2016;4:84. doi:10.1186/s40425-016-0193-2).
To address this further, the authors in this publication reviewed the efficacy of OPDIVO® in lung cancer patients with hepatic metastases, at their institution. A retrospective study was conducted and this analysis included data from 75 patients with advanced lung cancer, who were treated with PD1 inhibitor OPDIVO®, at East Carolina University. The study was designed to evaluate predictive markers of response to immune checkpoint blockade. Thirteen percent (13%) of the patients had liver metastases. The median age was 62 years, 22% of the patients had squamous cell, 33% had adenocarcinoma, and 44% had small cell neuroendocrine histology. Patients had an average of 1.7 therapies prior to treatment with OPDIVO®. Patients in this study received an average of 4 cycles of anti-PD1 therapy with OPDIVOreg;. Forty four percent (44%) of the patients received adjunctive therapy such as radiation (33%) or immune modulating chemotherapy, with the aim of augmenting the effect of the anti-PD1 therapy.
It was noted that that none of the patients with hepatic metastases experienced an objective decrease in their liver metastases after treatment with OPDIVO®. These patients had an average survival of 132 days after initial treatment with OPDIVO®.
The authors concluded that this is the largest reported series evaluating lung cancer patients with hepatic metastases, who had been treated with PD-1 inhibitors. They noted that consistent with previously published studies, patients with liver metastases have poor outcomes with anti-PD1 therapy and the mechanisms underlying such resistance must be elucidated, so that more effective treatment combinations can be developed. Outcomes with immune checkpoint inhibitor use in lung cancer patients with hepatic metastases. Addepalli S, Chipman R, Stroud G, et al. J Clin Oncol 35, 2017 (suppl 7S; abstract 38)
First Line KEYTRUDA® Superior to Chemotherapy in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Non Small Cell Lung Cancer accounts for approximately 85% of all lung cancers. The FDA in October, 2016 approved KEYTRUDA® (Pembrolizumab) for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), whose tumors have high PD-L1 expression (Tumor Proportion Score greater than or equal to 50%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and no prior systemic chemotherapy treatment for metastatic NSCLC.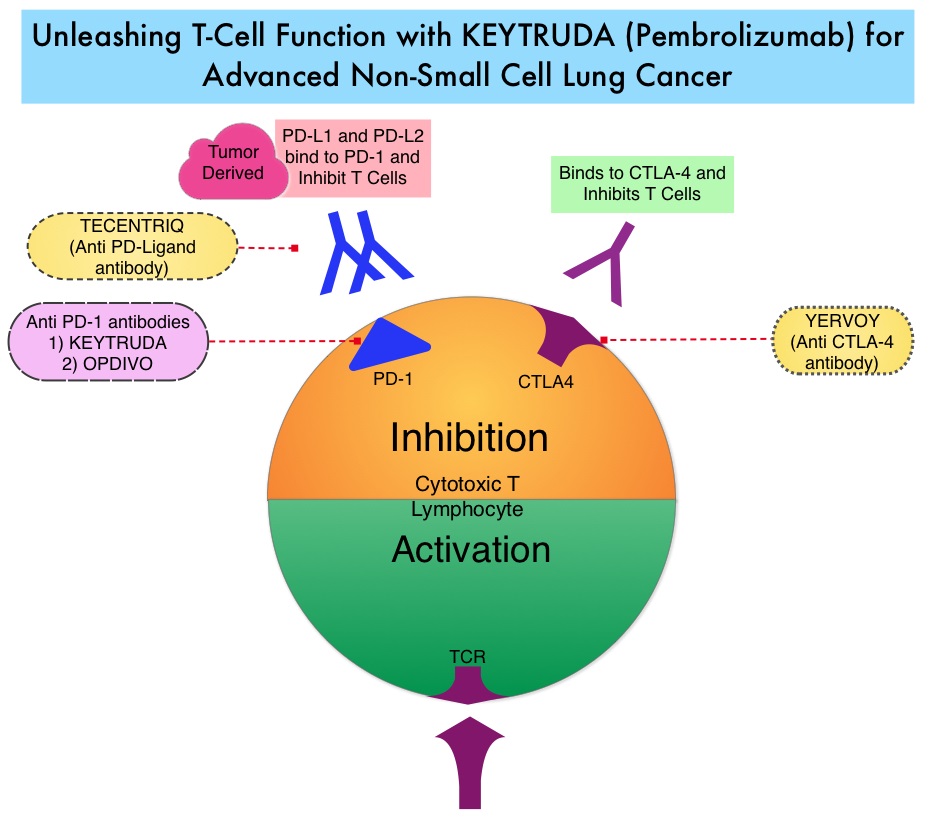
KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced Non Small Cell Lung Cancer (NSCLC) have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.
KEYNOTE-024 is an open-label, randomized, phase III trial in which KEYTRUDA® administered at a fixed dose was compared with investigator’s choice of cytotoxic chemotherapy, as first line therapy, for patients with advanced NSCLC, with tumor PD-L1 expression of 50% or greater. Three hundred and five (N=305) treatment naïve patients with advanced NSCLC and PD-L1 expression on at least 50% of tumor cells, were randomly assigned in a 1:1 ratio to receive either KEYTRUDA® (N=154) or chemotherapy (N=151). Enrolled patients had no sensitizing EGFR mutations or ALK translocations. Treatment consisted of KEYTRUDA® administered at a fixed dose of 200 mg IV every 3 weeks for 35 cycles or the investigator’s choice of platinum-based chemotherapy for 4-6 cycles. Pemetrexed (ALIMTA®) based therapy was permitted only for patients who had non-squamous tumors and these patients could receive ALIMTA® maintenance therapy after the completion of combination chemotherapy. The primary end point was Progression Free Survival and secondary end points included Overall Survival, Objective Response Rate and safety.
The median PFS was 10.3 months in the KEYTRUDA® group versus 6.0 months in the chemotherapy group (HR=0.50; P<0.001). This benefit was observed across all patient subgroups including tumor histologic type and chemotherapy regimen administered. The estimated Overall Survival at 6 months was 80.2% in the KEYTRUDA® group versus 72.4% in the chemotherapy group (HR=0.60; P=0.005). Patients in the KEYTRUDA® group experienced higher Response Rates than in the chemotherapy group (44.8% vs. 27.8%) as well as longer median duration of response (Not Reached versus 6.3 months). These benefits were realized even after 43.7% of the patients in the chemotherapy group following progression, had crossed over to receive KEYTRUDA®. Adverse events of any grade were less frequent in the KEYTRUDA® group compared to the chemotherapy group, with diarrhea, fatigue and pyrexia being more common in the KEYTRUDA® group whereas anemia, nausea and fatigue were more often noted in the chemotherapy group. As expected, immune-mediated adverse events (including pneumonitis) occurred more frequently with KEYTRUDA® whereas cytopenias occurred more frequently with chemotherapy.
It was concluded that in treatment naïve patients with advanced NSCLC and a PD-L1 tumor proportion score of 50% or greater, KEYTRUDA® was associated with significantly longer Progression Free and Overall Survival and with fewer adverse events, compared with platinum-based chemotherapy. This landmark trial is practice changing for advanced NSCLC. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. Reck M, Rodríguez-Abreu D, Robinson AG, et al. for the KEYNOTE-024 Investigators. October 9, 2016DOI: 10.1056/NEJMoa1606774
FDA Approves TECENTRIQ® for Non-Small Cell Lung Cancer
SUMMARY: The FDA on October 18, 2016, approved TECENTRIQ® (Atezolizumab) for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) whose disease progressed during or following platinum-containing chemotherapy. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2016 about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. The treatment paradigm for malignancies has been rapidly evolving, with a better understanding of the Immune checkpoints or gate keepers. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, Immune checkpoints or gate keepers inhibit an intense immune response by switching off the T cells of the immune system. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are now available that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), as well as Programmed cell Death Ligands (PD-L1), that are expressed by cells in the tumor micro environment. By targeting the Immune check point proteins or their ligands, T cells are unleashed, resulting in T cell proliferation, activation and a therapeutic response.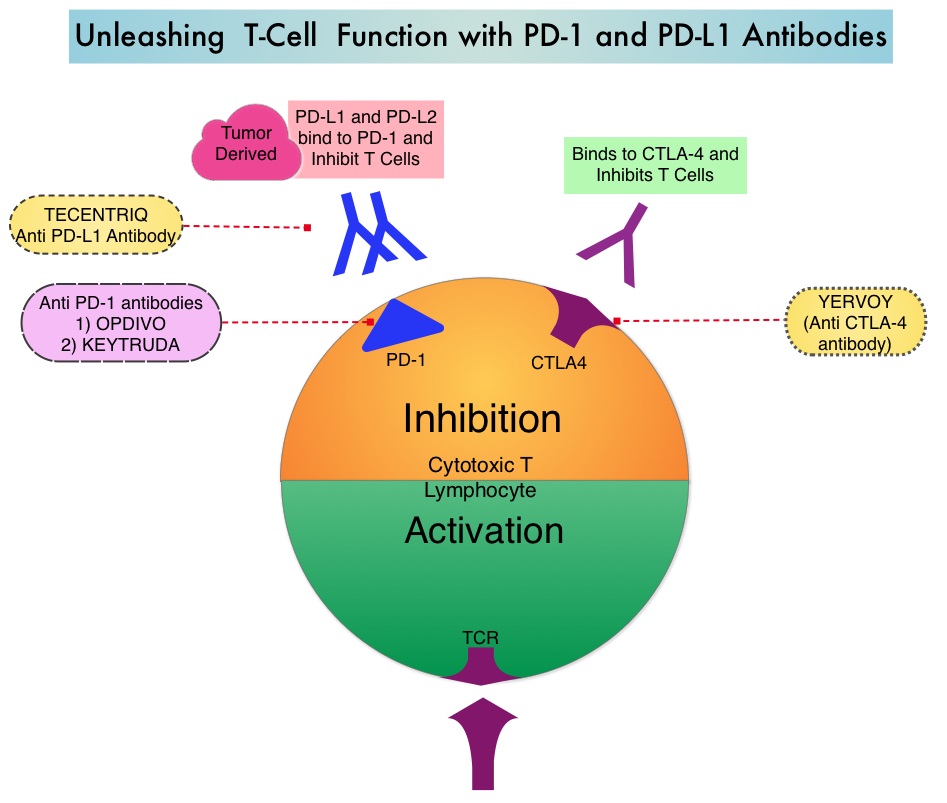
TECENTRIQ® (Atezolizumab) is an anti PD-L1 monoclonal antibody designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating Immune Cells, thereby blocking its interactions with PD-1 and B7.1 receptors and thus enabling the activation of T cells and restoring tumor-specific T-cell immunity. The approval of TECENTRIQ® was based two international, clinical trials (OAK and POPLAR trials). TECENTRIQ® demonstrated survival benefit compared to Docetaxel in a multicenter, randomized, phase II study (POPLAR trial). In this study, the median Overall Survival was 12.6 months and 9.7 months (HR=0.69) for the TECENTRIQ® and Docetaxel groups respectively.
OAK trial is a global, multicentre, open-label, randomized, controlled Phase III study in which 1225 patients with locally advanced or metastatic NSCLC, whose disease had progressed following previous treatment with platinum-containing chemotherapy, were enrolled. Patients with both squamous and non-squamous histology were randomized in a 1:1 ratio to receive either TECENTRIQ® 1200 mg IV every 3 weeks or Docetaxel 75 mg/m2 IV every 3 weeks. Patients were stratified according to PD-L1 status, number of prior chemotherapy regimens and histology. The median age was 64 years, 25% had 2 prior lines of therapy and 26% had squamous histology. The co-primary endpoints were Overall Survival (OS) in all randomized patients and in a PD-L1 selected subgroup in the primary analysis population. Secondary endpoints included Progression Free Survival, Objective Response Rate, Duration of Response and Safety.
The primary efficacy analysis was conducted and reported in the first 850 of 1225 total enrolled patients. The median Overall Survival was 13.8 months in the TECENTRIQ® group compared to 9.6 months in the Docetaxel group (HR=0.74; P=0.0004), with a 26% improvement in Overall Survival in the patient group who received TECENTRIQ®. This benefit was seen regardless of their PD-L1 expression levels, including patients whose tumors displayed PD-L1 expression of less than 1%. Patients with high PD-L1 expression had more pronounced benefit with TECENTRIQ® with a 59% improvement in OS compared with Docetaxel (HR=0.41; P<0.0001). The OS benefit was similar in patients with squamous or non-squamous histology. The most common adverse reactions in patients in patients treated with TECENTRIQ® were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation.
The authors concluded that TECENTRIQ® offers a new second-line therapeutic strategy for patients with Non Small Cell Lung Cancer, with Overall Survival benefit, regardless of the PD-L1 status of the tumor. Primary analysis from OAK, a randomized phase III study comparing atezolizumab with docetaxel in 2L/3L NSCLC. Barlesi F, Park K, Ciardiello F et al. Abstract LBA44_PR. Presented at: 2016 ESMO Congress; October 7–11 (2016) Copenhagen, Denmark.
FDA Approves KEYTRUDA® for Treatment Naïve Patients with Advanced NSCLC
SUMMARY: The FDA on October 24, 2016 approved KEYTRUDA® (Pembrolizumab) for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), whose tumors have high PD-L1 expression (Tumor Proportion Score greater than or equal to 50%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and no prior systemic chemotherapy treatment for metastatic NSCLC. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2016 about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer accounts for approximately 85% of all lung cancers.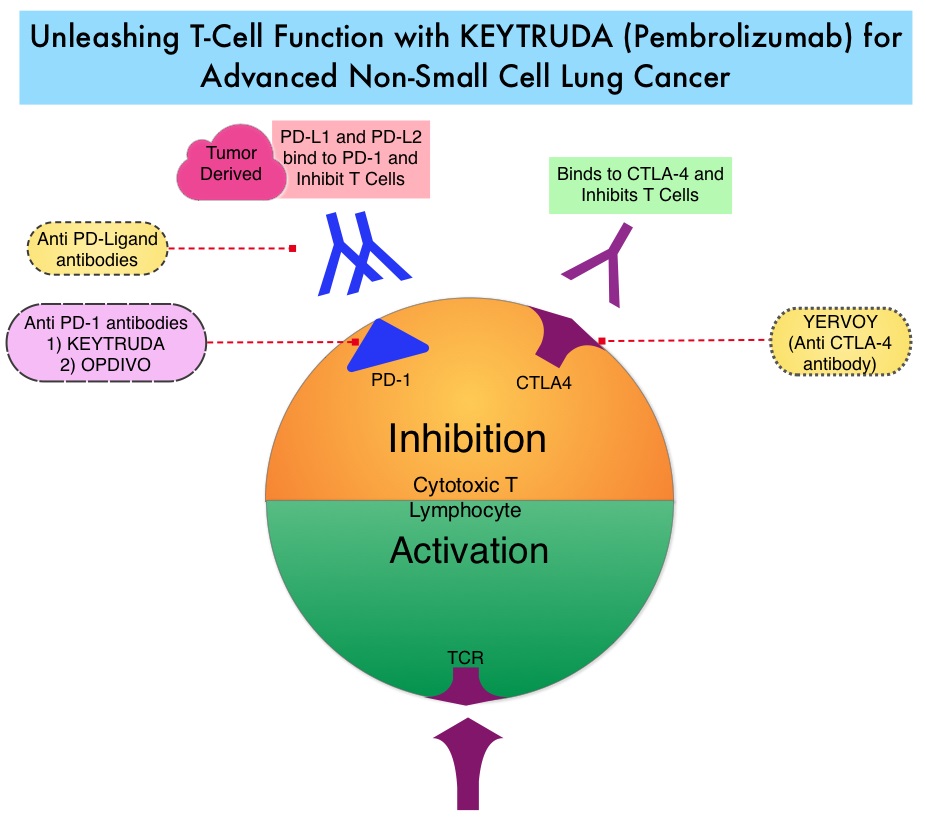
KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced Non Small Cell Lung Cancer (NSCLC) have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.
KEYNOTE-024 is a open-label, randomized, phase III trial in which KEYTRUDA® administered at a fixed dose was compared with investigator’s choice of cytotoxic chemotherapy, as first line therapy, for patients with advanced NSCLC, with tumor PD-L1 expression of 50% or greater. Three hundred and five (N=305) treatment naïve patients with advanced NSCLC and PD-L1 expression on at least 50% of tumor cells, were randomly assigned in a 1:1 ratio to receive either KEYTRUDA® (N=154) or chemotherapy (N=151). Enrolled patients had no sensitizing EGFR mutations or ALK translocations. Treatment consisted of KEYTRUDA® administered at a fixed dose of 200 mg IV every 3 weeks for 35 cycles or the investigator’s choice of platinum-based chemotherapy for 4-6 cycles. Pemetrexed (ALIMTA®) based therapy was permitted only for patients who had non-squamous tumors and these patients could receive ALIMTA® maintenance therapy after the completion of combination chemotherapy. The primary end point was Progression Free Survival and secondary end points included Overall Survival, Objective Response Rate and safety.
The median PFS was 10.3 months in the KEYTRUDA® group versus 6.0 months in the chemotherapy group (HR=0.50; P<0.001). This benefit was observed across all patient subgroups including tumor histologic type and chemotherapy regimen administered. The estimated Overall Survival at 6 months was 80.2% in the KEYTRUDA® group versus 72.4% in the chemotherapy group (HR=0.60; P=0.005). Patients in the KEYTRUDA® group experienced higher Response Rates than in the chemotherapy group (44.8% vs. 27.8%) as well as longer median duration of response (Not Reached versus 6.3 months). These benefits were realized even after 43.7% of the patients in the chemotherapy group following progression, had crossed over to receive KEYTRUDA®. Adverse events of any grade were less frequent in the KEYTRUDA® group compared to the chemotherapy group, with diarrhea, fatigue and pyrexia being more common in the KEYTRUDA® group whereas anemia, nausea and fatigue were more often noted in the chemotherapy group. As expected, immune-mediated adverse events (including pneumonitis) occurred more frequently with KEYTRUDA® whereas cytopenias occurred more frequently with chemotherapy.
It was concluded that in treatment naïve patients with advanced NSCLC and a PD-L1 tumor proportion score of 50% or greater, KEYTRUDA® was associated with significantly longer Progression Free and Overall Survival and with fewer adverse events, compared with platinum-based chemotherapy. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. Reck M, Rodríguez-Abreu D, Robinson AG, et al. for the KEYNOTE-024 Investigators. October 9, 2016DOI: 10.1056/NEJMoa1606774
FDA Approves TAGRISSO® (Osimertinib) Blood-Based T790M Companion Diagnostic Test
SUMMARY: The FDA on September 29, 2016 approved a blood-based companion diagnostic for TAGRISSO® (Osimertinib). The companion diagnostic for TAGRISSO® is the only FDA approved and clinically validated companion diagnostic test that uses either tissue or a blood sample to confirm the presence of a T790M point mutation in patients with metastatic Epidermal Growth Factor Receptor (EGFR) mutation-positive Non Small Cell Lung Cancer (NSCLC), who have had progression of disease on or after EGFR Tyrosine Kinase Inhibitor therapy. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2016 about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. Approximately 10% to 15% of Caucasian patients and 50% of Asian patients with Adenocarcinomas, harbor activating EGFR mutations and 90% of these mutations are either Exon 19 deletions or L858R point mutations in Exon 21. EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60% to 70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9 to 14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50% - 60% of patients.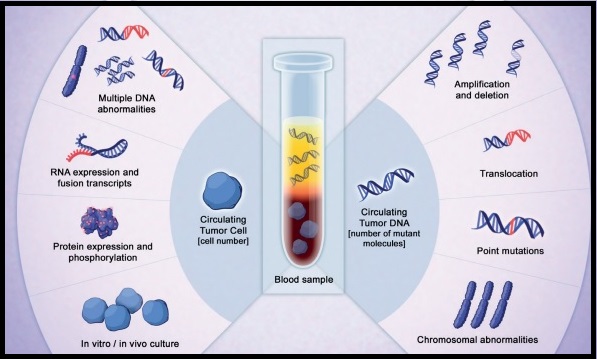
TAGRISSO® is presently approved by the FDA for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, who had progressed on prior systemic therapy, including an EGFR TKI. The application of precision medicine with targeted therapy requires detection of molecular abnormalities in a tumor specimen, following progression or recurrence. Archived biopsy specimens may not be helpful as it is important to identify additional mutations in the tumor at the time of recurrence or progression, in order to plan appropriate therapy. Further, recurrent tumors may be inaccessible for a safe biopsy procedure or the clinical condition of the patient may not permit a repeat biopsy. Additionally, the biopsy itself may be subject to sampling error due to tumor heterogeneity. Genotyping circulating-free tumor DNA (cfDNA) in the plasma can potentially overcome the shortcomings of repeat biopsies and tissue genotyping, allowing the detection of many more targetable gene mutations, thus resulting in better evaluation of the tumor genome landscape.
The COBAS® Mutation Test v2, is a real-time PCR test for the qualitative detection of defined mutations of the EGFR gene in NSCLC patients. Defined EGFR mutations are detected using DNA isolated from Formalin-Fixed Paraffin-Embedded Tumor tissue (FFPET) or circulating-free tumor DNA (cfDNA) from plasma, obtained from EDTA anti-coagulated peripheral whole blood (purple top tube). This new blood-based companion diagnostic test offers an important option to identify T790M mutation in patients with metastatic EGFR mutation-positive NSCLC, who have progressed on an EGFR TKI therapy, and for whom a tissue biopsy may not be feasible.
US FDA approves Tagrisso (osimertinib) blood-based T790M companion diagnostic test. AstraZeneca website. https://www.astrazeneca-us.com/content/az-us/media/press-releases/2016/us-fda-approves-tagrisso-osimertinib-blood-based-t790m-companion-diagnostic-test-09292016.html. Updated September 29, 2016.
Proton Beam Therapy May Improve Survival Compared to Conventional Radiation in Stage II and III NSCLC Patients
SUMMARY: Lung cancer is the second most common cancer in both men and women and the American Cancer Society estimates that for 2016 about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Even though Photon-based external beam radiation plus concurrent chemotherapy is the current standard of care for patients with unresectable stage III NSCLC, Proton beam therapy is emerging as an alternative to conventional Photon beam therapy for many cancer types. Radiation Therapy involves the use of X-Rays, Gamma rays and charged particles for cancer treatment. External beam radiation therapy is most often delivered using a linear accelerator in the form of Photon beams (either X-rays or Gamma rays). Photons have no mass and are packets of energy of an electromagnetic wave. Electrons and Protons are charged particles and Electrons are considered light particles whereas Protons are considered heavy particles. Electron beams are used to irradiate skin and superficial tumors, as they are unable to penetrate deep into the tissues. The different types of external beam radiation treatments include 3-Dimensional Conformal Radiation Therapy (3D-CRT) meant to deliver radiation to very precisely shaped target areas, IMRT or Intensity Modulated Radiation Therapy which allows different areas of a tumor or nearby tissues to receive different doses of radiation, Image Guided Radiation Therapy (IGRT) which allows reduction in the planned volume of tissue to be treated as changes in a tumor size are noted during treatment, Stereotactic RadioSurgery (SRS) which can deliver one or more high doses of radiation to a small tumor, Stereotactic Body Radiation Therapy (SBRT) or CYBERKNIFE® which is similar to SRS but also takes the normal motion of the body into account while treating malignancies involving the lung and liver and Proton beam therapy. Proton beams unlike Photons, enter the skin and travel through the tissues and deposit much of their energy at the end of their path (known as the Bragg peak) and deposit less energy along the way. This is unlike Photons which deposit energy all along the path through the tissues and the deposited dose decreases with increasing depth. As a result, with Proton beam therapy, normal tissues are exposed to less radiation compared with Photons. Despite this advantage, tissue heterogeneity such as organ motion, tumor volume changes during treatment can have a significant negative impact on target coverage for Proton beam therapy and can result in damage to the surrounding tissues and potential complications.
It has remained unclear whether Proton beam therapy improves Overall Survival (OS) in patients with NSCLC. To address this question, the authors conducted a retrospective analysis using the National Cancer Data Base (NCDB) and analyzed outcomes and predictors associated with Proton beam therapy for NSCLC. This analysis included 140,383 patients with stage I to stage IV NSCLC, treated with thoracic radiation from 2004-2012, of whom 59% had stage II and III disease. Of these patients, 140,035 were treated with Photon beam therapy and 348 with Proton beam therapy. The median age was 68 yrs, 57% were males, 85% were Caucasian, 27% were treated at academic centers and 78% in metropolitan areas. To reduce treatment selection bias, propensity score matching method was implemented.
It was noted that patients were less likely to receive Proton beam therapy in community or comprehensive community centers compared to academic centers (P< 0.001). Further, patients who received Proton beam therapy were more likely to have a higher education and income. On multivariate analysis, it was noted that the risk for death was greater with use of Photon beam therapy compared to Proton beam therapy (HR=1.46; P<0.001). Among patients with stage II and III disease, 5 year OS was superior with Proton beam therapy compared with Photon beam therapy (22.3% versus 15%; P=0.01). Patients with stage II and III disease who received Photon beam therapy had worse OS both in multivariate (HR=1.19; P=0.06) and univariate (HR=1.23; P=0.02) analyses, compared with Proton beam therapy. Proton beam therapy was associated with better 5 year OS compared to Photon beam therapy (23% vs. 14%; P=0.02), on propensity matched analysis. The median OS was 11 months with Photon therapy compared to 19 months with Proton therapy.
The authors concluded that in this retrospective database analysis, thoracic radiation with Proton beam therapy was associated with better survival rates for patients with stage II and III NSCLC. An ongoing randomized phase III trial (NRG Oncology 1308) involving stage III NSCLC patients is evaluating if chemotherapy and Proton beam therapy is superior to chemotherapy and Photon beam therapy. National Cancer Data Base analysis of proton versus photon radiotherapy in non-small cell lung cancer (NSCLC). Behera M, OConnell KA, Liu Y, et al. J Clin Oncol 34, 2016 (suppl; abstr 8501)
FDA Approves GILOTRIF® for Squamous Cell Carcinoma of the Lung
SUMMARY: The FDA on April 15, 2016 approved GILOTRIF® (Afatinib) tablets for the treatment of patients with advanced Squamous Cell Carcinoma of the lung, whose disease has progressed after treatment with Platinum-based chemotherapy. Lung cancer is the second most common cancer in both men and women and the American Cancer Society estimates that for 2016, about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. Non Small Cell Lung Cancer patients with Squamous Cell histology have been a traditionally hard- to-treat, patient group, with less than 5% of patients with advanced SCC, surviving for five years or longer. Some of the advanced NSCLC tumors are dependent on the Epidermal Growth Factor Receptor (EGFR) for cell proliferation and survival, regardless of EGFR mutation status. TARCEVA® (Erlotinib) is a reversible EGFR Tyrosine Kinase Inhibitor and is presently approved by the FDA for the treatment of locally advanced or metastatic NSCLC, after failure of at least one prior chemotherapy regimen. GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. GILOTRIF® was approved by the FDA in July 2013, for the first line treatment of patients with metastatic NSCLC, whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations.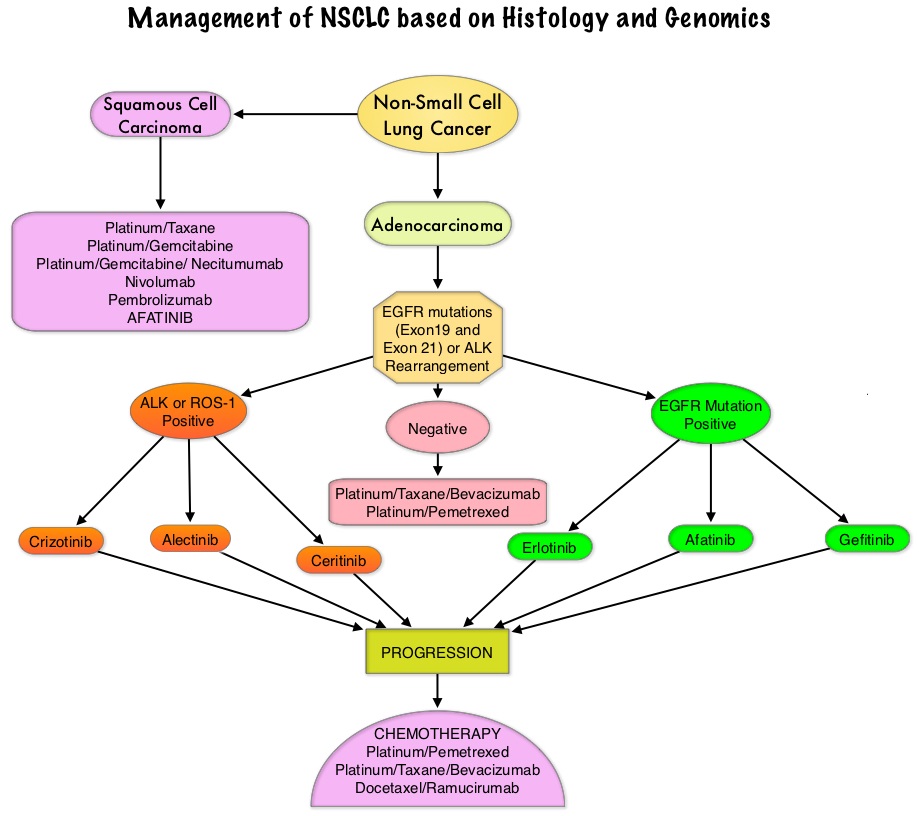
This additional indication approved by the FDA was based on the LUX-Lung 8 study, which is a phase III trial in which 795 patients with Stage IIIB/IV Squamous Cell Carcinoma of the lung who had progressed on first line platinum based doublet therapy, were randomized 1:1 to receive GILOTRIF® 40 mg PO daily (N=398) or TARCEVA® 150 mg PO daily (N=397). Treatment was given until disease progression. The median age was 65 years. Majority of the patients were male, caucasian and ex-smokers. The Primary endpoint was Progression Free Survival (PFS) and Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR), Disease Control Rate (DCR), patient reported outcomes and safety. The Primary endpoint of Progression Free Survival (PFS) was met and reported in 2014 and favored GILOTRIF® over TARCEVA®. The authors in this analysis reported the Overall Survival data, as well as updated data on Progression Free Survival and other Secondary endpoints. The median Overall Survival was 7.9 months with GILOTRIF® and 6.8 months with TARCEVA® (HR=0.81; P=0.007). This meant a 19% reduction in the risk of death with GILOTRIF® when compared to TARCEVA® and this survival advantage was consistent across all time points. The updated median Progression Free Survival for GILOTRIF® was 2.4 months versus 1.9 months for TARCEVA® (HR=0.82; P=0.04) which meant an 18% reduction in the disease progression. The Disease Control Rate was 51% for GILOTRIF® and 40% with TARCEVA® (P=0.002). Based on patient reported outcomes, symptoms including cough and dyspnea were better with GILOTRIF® compared to TARCEVA®. Incidence of severe adverse events was similar with both therapies, with patients on GILOTRIF® experiencing more grade 3 diarrhea and stomatitis and patients receiving TARCEVA® experiencing more grade 3 rash.
The authors concluded that GILOTRIF® should be the TKI of choice for the second line treatment of patients with Squamous Cell Carcinoma of the lung, as it significantly improves Overall Survival, Progression Free Survival, Disease Control Rate and controls symptoms with manageable toxicities, when compared to TARCEVA®. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Soria J, Felip E, Cobo M, et al. The Lancet Oncology 2015;16:897-907
Liquid Biopsy Can Rapidly Detect EGFR Mutations and KRAS mutations with High Specificity
SUMMARY: It has been well established that treatment with EGFR TKIs results in superior outcomes, for patients with tumors harboring exon 19 deletions and exon 21 mutations. The application of precision medicine with targeted therapy, requires detection of molecular abnormalities in a tumor specimen, following progression or recurrence. Archived biopsy specimens may not be helpful as it is important to identify additional mutations in the tumor at the time of recurrence or progression, in order to plan appropriate therapy. Further, recurrent tumors may be inaccessible for a safe biopsy procedure or the clinical condition of the patient may not permit a repeat biopsy. Additionally, the biopsy itself may be subject to sampling error due to tumor heterogeneity. Genotyping cell free DNA in the plasma can potentially overcome the shortcomings of repeat biopsies and tissue genotyping, allowing the detection of many more targetable gene mutations, thus resulting in better evaluation of the tumor genome landscape.
The purpose of this study was to prospectively validate plasma droplet digital PCR (ddPCR) for the rapid detection of common Epidermal Growth Factor Receptor (EGFR) and KRAS mutations, as well as the EGFR T790M acquired resistance mutation. The authors prospectively evaluated the feasibility and accuracy of this assay in patients with newly diagnosed advanced non-squamous Non Small Cell Lung Cancer (NSCLC) who either were newly diagnosed and initial therapy was planned (N=120) or had developed acquired resistance to an EGFR kinase inhibitor and rebiopsy was planned (N=60). The median age was 62 years and 62% were females.
Following initial blood sampling of all patients, plasma droplet digital Polymerase Chain Reaction (ddPCR) for EGFR and KRAS mutations, including EGFR exon 19 deletion, EGFR L858R, KRAS G12X and EGFR T790M acquired resistance mutation, was performed. All patients underwent biopsy for tissue genotyping, and this was used as a reference standard for comparison with the liquid biopsy results. Important study outcomes included sensitivity and specificity of plasma ddPCR assay, as well as test turnaround time, which was defined as the number of business days between blood sampling and test reporting.
Tumor genotypes identified included 80 EGFR exon 19 or L858R mutations, 35 EGFR T790M mutations, and 25 KRAS G12X mutations. The ddPCR assay median turnaround time was 3 days compared with 12 days for tissue genotyping and 27 days for patients with acquired resistance. Plasma ddPCR exhibited a positive predictive value of 100% for EGFR 19 del, 100% for EGFR L858R mutation and 100% for KRAS. The positive predictive value for EGFR T790M was lower at 79%. The sensitivity of plasma ddPCR assay was 82% for EGFR exon19 del, 74% for EGFR L858R mutation, and 77% for EGFR T790M acquired resistance mutation, but lower for KRAS at 64%. Sensitivity for EGFR or KRAS was higher in patients with multiple metastatic sites (P=0.001), specifically in those with bone and hepatic metastases.
The authors concluded that in this first prospective study, plasma ddPCR assay can rapidly detect EGFR and KRAS mutations with high specificity, allowing treatment selection, without repeat biopsies. Additionally, this assay may also detect EGFR T790M mutation, missed by tissue genotyping, due to tumor heterogeneity in resistant disease. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. Sacher AG, Paweletz C, Dahlberg SE, et al. JAMA Oncol. Published online April 07, 2016. doi:10.1001/jamaoncol.2016.0173
FDA Approves XALKORI® for ROS1-Rearranged Non Small Cell Lung Cancer
SUMMARY: The FDA on March 11, 2016, approved XALKORI® (Crizotinib) for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), whose tumors are ROS1-positive. XALKORI® was first approved in 2011 for the treatment of patients with NSCLC, whose tumors are Anaplastic Lymphoma Kinase (ALK) positive. Lung cancer is the second most common cancer in both men and women and the American Cancer Society estimates that for 2016, about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous cell carcinomas, 40% are Adenocarcinomas and 10% are Large cell carcinomas. There is now growing body of evidence suggesting superior outcomes when advanced NSCLC patients with specific genomic alterations receive targeted therapies. Approximately 1-2% of lung adenocarcinomas harbor ROS1 gene rearrangements. ROS1 gene is located on chromosome 6q22 (long arm of chromosome 6) and plays an important role in cell growth and development. ROS1 gene fusion with another gene results in a mutated DNA sequence which then produces an abnormal protein responsible for unregulated cell growth and cancer. ROS1 gene rearrangement has been identified as a driver mutation in Non Small Cell Lung Cancer with adenocarcinoma histology. This is more common in nonsmokers or in light smokers (<10 pack years), who are relatively young (average age of 50 years) and thus share similar characteristics with ALK-positive patients. The ROS protein and the ALK protein have similar structure and function and are sensitive to Tyrosine Kinase Inhibitors such as XALKORI® (Crizotinib) and ZYKADIA® (Ceritinib). ROS1 mutations have been also been associated with Cholangiocarcinoma (Bile duct cancer) and Glioblastoma multiforme. ROS1 rearrangements are mutually exclusive with other oncogenic mutations found in NSCLC such as EGFR mutations, KRAS mutations and ALK rearrangement. The presence of a ROS1 rearrangement can be detected by Fluorescence In Situ Hybridization (FISH), ImmunoHistoChemistry (IHC), Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) and Next Generation-Sequencing. XALKORI® is a small molecule Tyrosine Kinase Inhibitor that targets ALK, MET and ROS1 tyrosine kinases.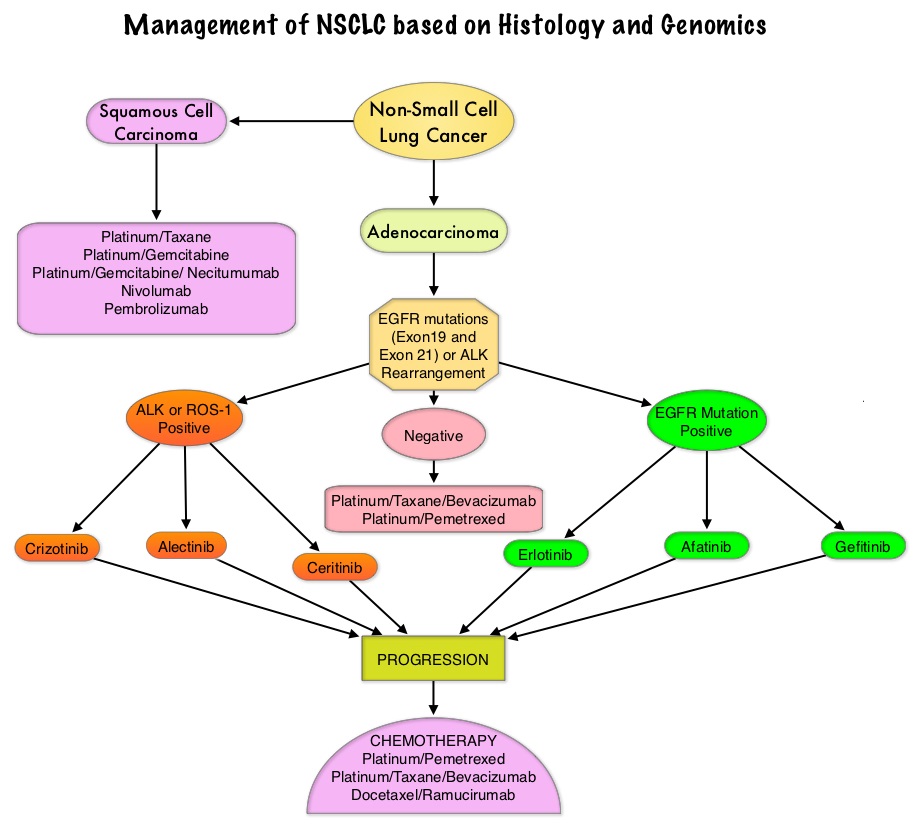
The latest FDA approval was based on the results of a multicenter, single-arm, expansion cohort of the phase I study of XALKORI®, in which 50 patients with advanced NSCLC, who tested positive for ROS1 rearrangement, were enrolled. The median age was 53 years, 98% had adenocarcinoma histology and majority of patients (86%) had received previous treatment for advanced disease, with 44% having received more than 1 prior therapy. XALKORI® was administered orally at 250 mg twice daily in continuous 28-day cycles. Treatment was continued until disease progression or unacceptable toxicities. The primary end point was Objective Response Rate, and Duration of Response (DoR) was an additional outcome measure.
The Objective Response Rate by investigator assessment was 72%, with 3 Complete Responses and 33 Partial Responses. The median Duration of Response was 17.6 months. The median Progression Free Survival was 19.2 months and Overall Survival rate at 12 months was 85%. The most common adverse reactions associated with XALKORI® were vision disorders, nausea, diarrhea, vomiting, edema, elevated transaminases, fatigue, upper respiratory infection and neuropathy.
The authors concluded that XALKORI® has significant antitumor activity in patients with advanced ROS1-rearranged NSCLC. The significantly superior median Duration of Response (17.6 vs 11.4 months) and median Progression Free Survival (19.2 vs 9.7 months) in the ROS1-rearranged NSCLC compared to ALK- rearranged NSCLC, may be due to more potent inhibition of ROS1 than ALK, by XALKORI®, resulting in more effective target inhibition and more durable responses. Crizotinib in ROS1-Rearranged Non–Small-Cell Lung Cancer. Shaw AT, Ou S-HI, Bang Y-J, et al. N Engl J Med 2014; 371:1963-1971
Lobectomy Superior to Sublobar Resection in Early Stage Non Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and the American Cancer Society estimates that for 2016 about 224,390 new cases of lung cancer will be diagnosed and over 158,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Lobectomy is the treatment of choice for resectable Non Small Cell Lung Cancer (NSCLC). Pneumonectomy is rarely performed due to unacceptably high mortality rate. Sublobar resection (Wedge resection or Segmentectomy) is considered a “compromise operation” in selected high risk patients with early stage lung cancer. With the approval of lung cancer screening in high risk individuals and subsequent detection of small tumors, Sublobar resections have been on the rise, even in good-risk patients, in many institutions. Sublobar resection includes Wedge resection and Segmentectomy. In Wedge resection, the lung tumor is removed with a surrounding margin of normal lung tissue, and is not an anatomical resection. Segmentectomy, unlike Wedge resection, is an anatomical resection that usually includes one or more pulmonary parenchymal segments with the dissection of intraparenchymal and hilar lymph nodes. Wedge resection is inferior to anatomic Segmentectomy and is associated with an increased risk of local recurrence and decreased survival in patients with Stage I NSCLC.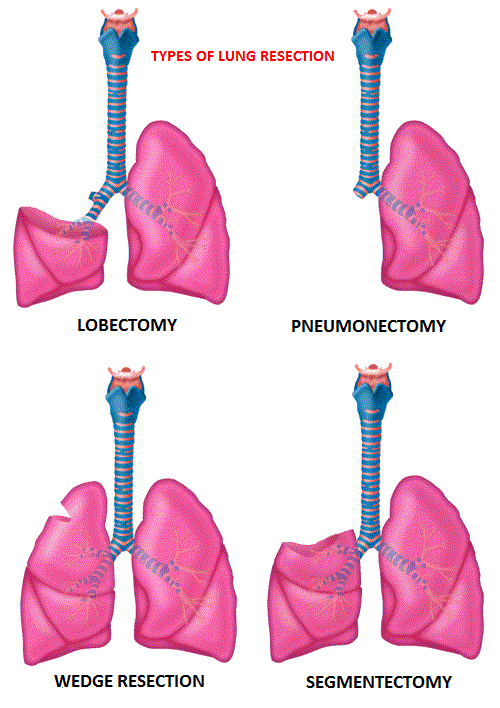
The authors in this study analyzed the National Cancer Data Base (NCDB) and the primary goal of this study was to understand practice patterns in the surgical management of patients with clinical Stage IA NSCLC, as well as identify predictors of surgical management with Sublobar resection versus Lobectomy and also evaluate the extent of pathologic lymph node assessment, performed in association with Sublobar resections, in a community practice setting. A secondary goal was to compare long term survival between Sublobar resection versus Lobectomy.
In this large analysis, 39,403 patients from the National Cancer Data Base (NCDB) were included, of whom 75.5% (N=29,736) underwent Lobectomy and 24.5% (N=9667) had Sublobar resection (Wedge resection 84.7%; N = 8192 and Segmental resection 15.3%; N = 1475). Lymph node evaluation was not performed in 2788 (28.8%) of Sublobar resection patients, and 7298 (75.5%) of Sublobar resections were for tumors ≤ 2 cm.
It was noted that Lobectomy was associated with significantly improved 5-year survival compared to Sublobar resection (66.2% vs. 51.2%; adjusted HR=0.66; P <0 .001). Among patients who underwent Sublobar resection, lymph node sampling was associated with significantly better 5-year survival compared to patients who did not have lymph node sampling (58.2% vs. 46.4%; P < 0.001), although these outcomes were still inferior to Lobectomy.
The authors concluded that for patients with Stage 1A NSCLC, surgical Lobectomy significantly improved survival compared to Sublobar resection. Patients ineligible for Lobectomy and treated with Sublobar resection, should undergo lymph node samplings to help guide appropriate post operative therapy. Sublobar Resection for Clinical Stage IA Non–small-cell Lung Cancer in the United States. Speicher PJ, Gu L, Gulack BC, et al. Clinical Lung Cancer 2016;17:47-55
FDA Approves ALECENSA® for Metastatic ALK-Positive Non Small Cell Lung Cancer
SUMMARY: The FDA on December 11, 2015 granted accelerated approval to Alectinib (ALECENSA®) for the treatment of patients with Anaplastic Lymphoma Kinase (ALK)-positive metastatic Non Small Cell Lung Cancer (NSCLC), who have progressed on or are intolerant to XALKORI® (Crizotinib). Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous cell carcinomas, 40% are Adenocarcinomas and 10% are Large cell carcinomas. The discovery of rearrangements of the Anaplastic Lymphoma Kinase (ALK) gene in some patients with advanced NSCLC and adenocarcinoma histology, led to the development of agents such as XALKORI® (Crizotinib) and ZYKADIA® (Ceritinib), with promising results. It has become clear that appropriate, molecularly targeted therapy for tumors with a molecular abnormality, results in the best outcomes. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8% as well as other mutations in BRAF, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive and the presence of two simultaneous mutations, are rare.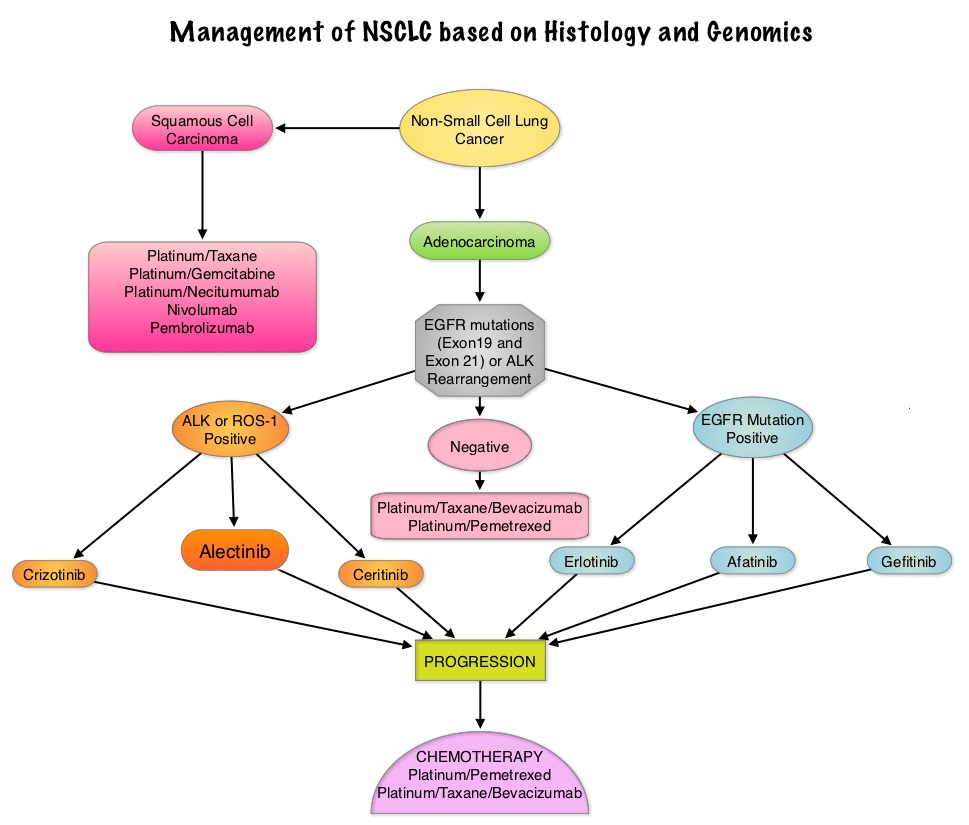
The approval of ALECENSA®) was based on two multicenter, single arm, open label, clinical trials (Study 1 and 2) in which enrolled patients received ALECENSA® 600 mg twice daily. The primary end point was Objective Response Rate (ORR). Secondary end points included Duration of Response (DoR), Objective Response Rate in the Central Nervous System (CNS) in those with measurable lesions in the CNS, and CNS Duration of Response. In Study 1 (N=87), the ORR was 38% and the Duration of Response was 7.5 months. In Study 2 (N=138), the ORR was 44% and the Duration of Response was 11.2 months. In a pooled analysis of patients from both Study 1 and Study 2 with measurable CNS lesions, the CNS Objective Response Rate was 61% and the median CNS Duration of Response was 9.1 months. The most common grade 1-2 adverse events were fatigue, constipation, edema, myalgia, anemia and elevation in liver function tests. The most common but rare grade 3-4 adverse reaction was dyspnea. It should be noted that ZYKADIA® (Ceritinib) is already approved for a similar patient population. A global phase III trial comparing ALECENSA® with XALKORI® as first line treatment, is presently underway.
A phase II, open-label, multicenter study of the ALK inhibitor alectinib in an ALK+ non-small-cell lung cancer (NSCLC) U.S./Canadian population who had progressed on crizotinib (NP28761). Gandhi L, Shaw A, Gadgeel SM, et al. J Clin Oncol 33, 2015 (suppl; abstr 8019)
Alectinib in Crizotinib-Refractory ALK-Rearranged Non–Small-Cell Lung Cancer: A Phase II Global Study. Ou SI, Ahn JS, Petris LD, et al. Published online before print November 23, 2015, doi: 10.1200/JCO.2015.63.9443
FDA Approves PORTRAZZA® for Metastatic Squamous Non-Small Cell Lung Cancer
SUMMARY: The FDA on November 24, 2015, granted approval to Necitumumab (PORTRAZZA®) in combination with GEMZAR® (Gemcitabine) and Cisplatin for the first line treatment of patients with metastatic Squamous Non Small Cell Lung Cancer (NSCLC). Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas.
Epidermal Growth Factor Receptor (EGFR) a Receptor Tyrosine Kinase (RTK) has been long known to control malignant cell proliferation, growth, survival, metabolism and migration. Therefore targeting EGFR with monoclonal antibodies has proven to be an effective strategy for the treatment of cancer. The two EGFR targeted monoclonal antibodies that have been available in the US include ERBITUX® (Cetuximab-chimeric IgG1) and VECTIBIX® (Panitumumab-human IgG2). PORTRAZZA® is a human IgG1 monoclonal antibody which also binds to the human Epidermal Growth Factor Receptor and blocks the binding of EGFR to its ligands.
The approval of PORTRAZZA® was based on the results of an open label, multicenter, multinational, phase III trial in which treatment naïve patients with metastatic Squamous NSCLC (N=1093) were randomized to receive PORTRAZZA® in combination with GEMZAR® (Gemcitabine) and Cisplatin (N=545) or GEMZAR® and Cisplatin alone (N=548). Treatment consisted of either PORTRAZZA® 800 mg IV days 1 and 8, GEMZAR® 1250 mg /m2 IV on days 1 and 8 along with Cisplatin 75mg/m2 IV on day 1 of each of a 21 day cycle or GEMZAR® and Cisplatin alone. Both treatment groups were well balanced and median age of patients was 62 years. The primary endpoint was Overall Survival and secondary endpoints included Progression Free Survival (PFS) and Overall Response Rate (ORR).
At a median follow up of 25 months, the median OS was 11.5 months in the PORTRAZZA® group and 9.9 months in the chemotherapy alone control group (HR = 0.84; P=0.01). The median PFS was 5.7 months in the PORTRAZZA® group and 5.5 months in the control group (HR=0.85; P=0.02). There was no difference in ORR noted in the two treatment groups (31% vs 29%). More patients in the PORTRAZZA® group experienced skin rash and hypomagnesemia and patients will therefore require close monitoring of serum electrolytes.
The authors concluded that the addition of PORTRAZZA® to GEMZAR® and Cisplatin chemotherapy significantly improves Overall Survival in patients with advanced Squamous NSCLC and represents a new first line treatment option for this malignancy. Because of the lack of benefit, PORTRAZZA® is not indicated for the treatment of non-Squamous NSCLC. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial. Thatcher N, Hirsch FR, Luft AV, et al. Lancet Oncol. 2015;16:763-774
FDA Approves TAGRISSO®, a Third Generation TKI, for EGFR T790M-Positive Non Small Cell Lung Cancer
SUMMARY: The U.S. FDA granted accelerated approval to TAGRISSO® (Osimertinib), for the treatment of patients with metastatic Epidermal Growth Factor Receptor (EGFR) T790M mutation-positive Non Small Cell Lung Cancer (NSCLC), as detected by an FDA-approved test, who had progressed on or after EGFR Tyrosine Kinase Inhibitor (TKI) therapy. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Approximately 10% to 15% of Caucasian patients and 50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R point mutations in Exon 21. EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60% to 70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9 to 14 months. This resistance to frontline EGFR TKI therapy has been attributed to acquired T790M “gatekeeper” point mutation in EGFR, identified in 50% - 60% of patients. The approval of TAGRISSO® was based on two multicenter, single arm, open label clinical trials (AURA and AURA2), in patients with metastatic EGFR T790M mutation-positive NSCLC, who had progressed on prior systemic therapy, including an EGFR TKI.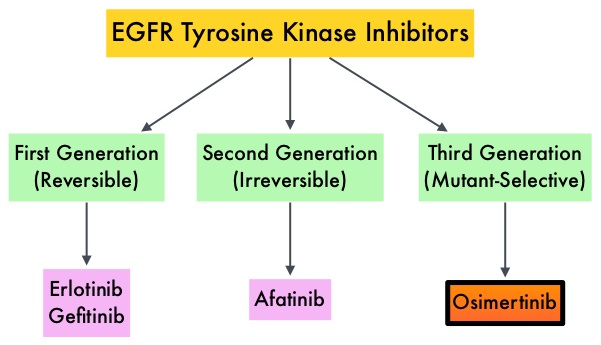
In the AURA dose escalation/expansion study (Study 1), 201 patients with EGFR mutation-positive advanced NSCLC received TAGRISSO® 80 mg PO daily until disease progression. Tumor samples were taken from all patients after disease progression on the most recent line of therapy, for prospective confirmation of T790M positive status by central laboratory testing, before enrollment. The median age was 62 years. The primary endpoint was Objective Response Rate (ORR) and secondary endpoints included Disease Control Rate (DCR), duration of response (DoR) and Progression Free Survival (PFS). The ORR in an updated analysis at the 2015 WCLC was 61% and DCR was 92%. The ORRs were similar across all lines of therapy, ie. Second line vs third line or more. The median DoR and median PFS have not been reached.
In the AURA2 Phase II study, 210 patients with locally advanced or metastatic NSCLC received TAGRISSO® 80 mg PO daily until disease progression. All eligible patients progressed on a previous EGFR TKI treatment and had a mandatory tumor sample taken after disease progression on the most recent line of therapy, for confirmation of T790M positive status by central laboratory testing. The median age was 64 years. The primary endpoint was Objective Response Rate (ORR) and secondary end points included Disease Control Rate (DCR), Duration of Response (DoR), Progression Free Survival (PFS), and safety. The ORR in an updated analysis presented at the 2015 WCLC was 71%, with 2 complete responses. The stable disease rate at 6 weeks or more was 21%, for a Disease Control Rate of 92%. The median Duration of Response was 7.8 months. The median Progression Free Survival (PFS) was 8.6 months.
Grade 1-2 toxicities from these two trials, which included a total of 411 patients included diarrhea, rash, dry skin, nail toxicity, eye disorders, nausea, decreased appetite and constipation. From these two studies it was concluded that TAGRISSO® is a new treatment option for patients who test positive for the EGFR resistance mutation, T790M, with significant response rates noted in over 50% of the treated patients. AZD9291 in pre-treated T790M positive advanced NSCLC: AURA2 Phase II study. Mitsudomi T, Tsai C, Shepherd F, et al. Presented at: 16th World Conference on Lung Cancer; September 6-9; Denver, CO. Abstract 1406.
FDA Approves KEYTRUDA® for Advanced Lung Cancer
SUMMARY: The FDA granted accelerated approval to KEYTRUDA® (Pembrolizumab), for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), whose tumors express Programmed Death Ligand 1 (PD-L1), as determined by an FDA-approved test, following disease progression on or after platinum-containing chemotherapy. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers.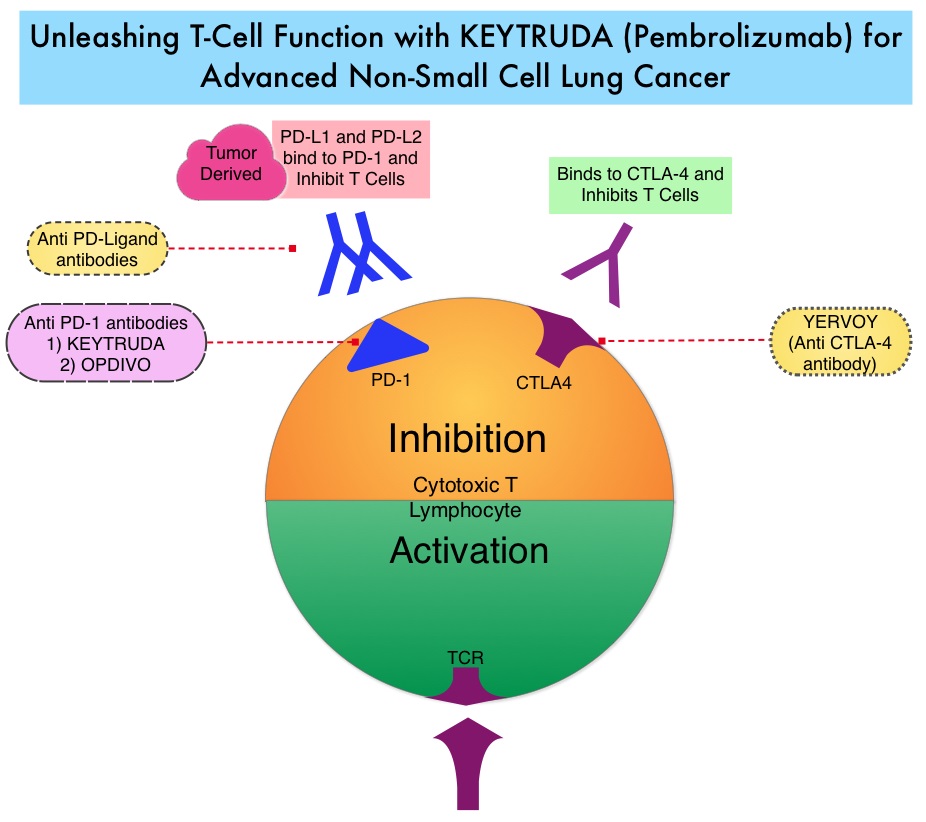
The treatment paradigm for solid tumors has been rapidly evolving with a better understanding of the Immune checkpoints. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, Immune checkpoints or gate keepers, switch off the T cells of the immune system and thereby inhibit an intense immune response. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies have been developed, that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4), also known as CD152, PD-1(Programmed cell Death-1), etc. Targeting Immune checkpoints unleashes the T cells, resulting in T cell proliferation, activation and a therapeutic response. KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells.
In this publication, the authors assessed the efficacy and safety of KEYTRUDA® in patients with advanced NSCLC enrolled in the KEYNOTE-001 phase I trial. Four Hundred and Ninety five (N=495) patients were assigned to either a training group (N=182) or a validation group (N=313) and KEYTRUDA® was administered at three dosages: 2 mg/kg IV every 3 weeks, 10 mg/kg IV every 3 weeks, or 10 mg/kg IV every 2 weeks. Patient responses were assessed every 9 weeks.
The Objective Response Rate (ORR) in the entire study population was 19.4%, the median Duration of Response was 12.5 months, the median Progression Free Survival was 3.7 months and the median Overall Survival was 12.0 months. The PD-L1 (Programmed Death-Ligand 1) expression was evaluated in 204 patients in the validation group by ImmunoHisto Chemistry (IHC) and membrane PD-L1 expression of 50% or more, in tumor cells, was selected as the cutoff. It was noted that among patients with PD-L1 expression in at least 50% of tumor cells, the Objective Response Rate was 45.2%, median Progression Free Survival was 6.3 months and median Overall Survival has not been reached. Responses were not as robust in those patients with tumors demonstrating less than 50% PD-L1 expression, but in those who did respond, the duration of responses were comparable to those with 50% or more PD-L1 expression. KEYTRUDA® was well tolerated overall and the common immune mediated adverse events were infusion reactions, hypothyroidism and pneumonitis.
The authors concluded that KEYTRUDA® showed significant antitumor activity in patients with advanced Non Small Cell Lung Cancer, whose tumor PD-L1expression was 50% or more. Further, the median duration of response exceeded 12 months among responders, regardless of the degree of PD-L1 expression. This study validated that PD-L1 expression in tumors is clearly a marker of response to KEYTRUDA®. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. Garon EB, Rizvi NA, Hui R, et al. N Engl J Med 2015; 372:2018-2028
Stereotactic Body Radiation Therapy (SBRT) Instead of Surgery for Patients with Early Stage Inoperable or Advanced Oligometastatic NSCLC
SUMMARY: Stereotactic RadioSurgery (SRS) is a non-surgical procedure that allows delivery of significantly higher doses of precisely focused radiation to the tumor, compared to conventional radiation therapy, with less collateral damage to the surrounding normal tissue. The technologies used for SRS include GAMMA KNIFE® which uses highly focused gamma rays, Proton Beam therapy which uses ionized hydrogen or Protons, Linear Accelerator (LINAC) and CYBER KNIFE® which use Photons, to target the tumor tissue. Stereotactic Body Radiation Therapy (SBRT) refers to stereotactically guided radiation therapy delivered over several days. Because SBRT is fractionated and is offered in three precise treatments, the short-and long-term side effects of radiation therapy are decreased and may allow higher total dosage to be given.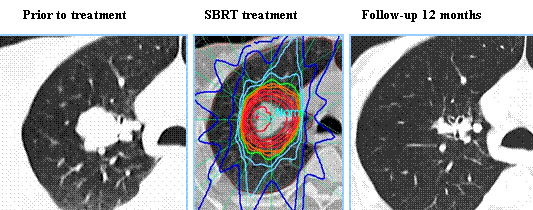
SBRT is a viable option for elderly and frail patients and those with comorbidities or those who decline surgery. Two studies presented at the 56th Annual Meeting of the American Society for Radiation Oncology (ASTRO) have provided convincing evidence in favor of SBRT in patients with inoperable early-stage lung cancer and for patients with oligometastatic stage IV Non Small Cell Lung Cancer (NSCLC). RTOG 0236 is a phase II trial in which 59 frail, elderly patients with early stage, medically inoperable Stage I Non Small Cell Lung Cancer received SBRT in three fractions of 18 Gy (total of 54 Gy) over a period of 10 days to 2 weeks. The median age was 72 years and these patients had multiple comorbidities that precluded them from curative surgery. The primary end point was 2-year actuarial primary tumor control. Secondary end points included Disease Free Survival (i.e., primary tumor, involved lobe, regional, and disseminated recurrence), treatment-related toxicity and Overall Survival. At 5 years, the Disease Free Survival and Overall Survival were 26% and 40%, respectively and the median Overall Survival was 4 years. The 5-year primary tumor and involved lobe (local) failure rate was 20%, local-regional failure rate was 38% and disseminated failure rate was 31%. In a second study, Ashworth and colleagues reported the individual patient data meta-analysis, which included 757 patients diagnosed with stage IV NSCLC at 20 cancer centers worldwide. All patients had 1-5 synchronous or metachronous metastases treated with surgical metastectomy, SBRT, or radical external beam radiation therapy and the primary tumor was treated aggressively with a curative intent. The 1-year Overall Survival (OS) was 70.2% and 5-year Overall Survival was 29.4%. The authors were able to develop a risk stratification model for survival, to help identify which patients would be the best candidates for SBRT or surgery. They noted that patients with metachronous metastases had a 5-year Overall Survival of 48% and were considered low risk, those with synchronous metastases and negative nodes had a 5-year OS of 36% and were considered intermediate risk and patients with synchronous metastases and positive nodes were considered high risk and had a 5-year overall survival of 14%.
Taken together, these two studies have demonstrated that SBRT improves Overall Survival in elderly frail patients with medically inoperable early stage Lung Cancer and SBRT also improves Overall survival in patients Stage IV Non Small Cell Lung Cancer, with metachronous metastases without nodal involvement. A multidisciplinary team approach is strongly recommended, as treatment decisions are made for the latter group.
1)Long-term Results of RTOG 0236: A Phase II Trial of Stereotactic Body Radiation Therapy (SBRT) in the Treatment of Patients with Medically Inoperable Stage I Non-Small Cell Lung Cancer. Timmerman RD, Hu C, Michalski J, et al. DOI:http://dx.doi.org/10.1016/j.ijrobp.2014.05.135
2)An Individual Patient Data Meta-Analysis of Outcomes and Prognostic Factors After Treatment of Oligometastatic Non-Small Cell Lung Cancer. Ashworth A, Senan S, Palma DA, et al. DOI: http://dx.doi.org/10.1016/j.ijrobp.2014.08.028
The FDA approves IRESSA® for metastatic Non Small Cell Lung Cancer
SUMMARY: The FDA on July 13, 2015 approved IRESSA® (Gefitinib) for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC), whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations, as detected by an FDA approved test. IRESSA was approved concurrently with a labeling expansion of the therascreen EGFR RGQ PCR Kit, a companion diagnostic test, for patient selection. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Epidermal Growth Factor Receptor (EGFR) is frequently overexpressed in NSCLC. In 2004, the discovery of Epidermal Growth Factor Receptor (EGFR) mutations in some advanced Non Small Cell Lung Cancer (NSCLC) patients, with Adenocarcinoma histology, and the favorable responses with EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), has changed the treatment paradigm, in favor of targeted therapy, for this patient subset. GILOTRIF® is an irreversible blocker of the ErbB family, which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. It is estimated that approximately 10% of Western patient population and 50% of Asian patients with NSCLC, harbor EGFR activating mutations. IRESSA® is an oral, EGFR Tyrosine Kinase Inhibitor (TKI), which works by blocking the activity of the EGFR tyrosine kinase enzyme responsible for regulating signaling pathways, implicated in the growth and survival of cancer cells. IRESSA® was granted Orphan Drug Designation by the FDA in August 2014 for the treatment of EGFR mutation positive NSCLC.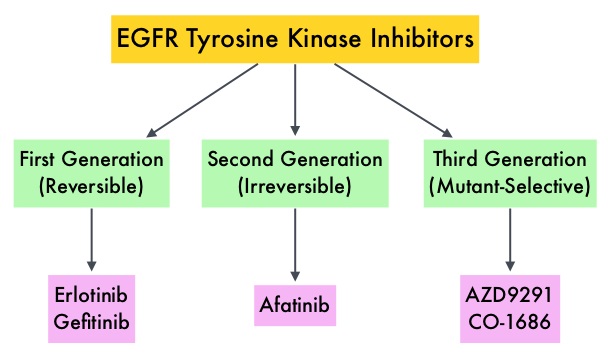
The approval of IRESSA® was based on the results of a Phase IV, single-arm, multicenter, open-label clinical study (IRESSA Follow-Up Measure or IFUM study) which included 106 treatment naïve-patients with metastatic EGFR mutation positive NSCLC who received IRESSA® 250mg PO daily. Treatment was given until disease progression or intolerable toxicity. Primary endpoint was Objective Response Rate (ORR). Secondary endpoints included Disease Control Rate (DCR), Progression Free Survival (PFS), Overall Survival (OS) and safety/tolerability. At the time of data cutoff, the investigator determined ORR was 70%, Duration of Response was 8.3 months, Disease Control Rate was 90.6%, median PFS was 9.7 months and median OS was19.2 months. This efficacy data was further supported by the IRESSA Pan-ASia Study (IPASS), a randomized phase III trial, which enrolled 1,217 treatment naïve advanced NSCLC patients with adenocarcinoma histology. Patients were randomized (1:1) to receive IRESSA® 250 mg PO daily or up to 6 cycles of combination chemotherapy with Carboplatin and Paclitaxel. The efficacy outcomes included Progression Free Survival (PFS) and Objective Response Rate (ORR). An exploratory analysis of a subset of 186 of 1217 patients (15%), who were determined to be EGFR mutation positive, had imaging studies available for evaluation (IRESSA® treated patients=88 and Carboplatin/Paclitaxel treated patients=98). The median PFS in the IRESSA® treated group was 10.9 months compared to 7.4 months for the Carboplatin/Paclitaxel treated patients (HR=0.54). The ORR was 67% with a Duration of Response (DoR) of 9.6 months for IRESSA® treated patients versus 41%, with a DoR of 5.5 months for Carboplatin/Paclitaxel treated patients. The most commonly reported adverse events for IRESSA® were diarrhea and skin toxicities including rash, acne, dry skin and pruritus. It was concluded that EGFR mutations are the strongest predictive biomarker for Progression Free Survival and tumor response to first line treatment with IRESSA®. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Douillard J-Y, Ostoros G, Cobo M, et al. Br J Cancer. 2014;110:55–62
GILOTRIF® Superior to TARCEVA® in Squamous Cell Carcinoma of the Lung
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas.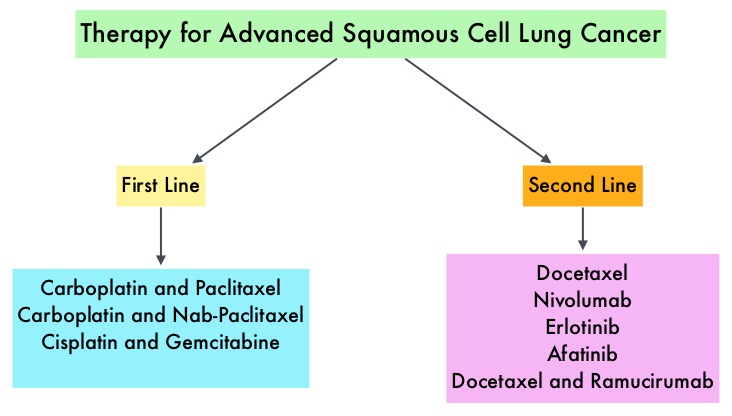 Non Small Cell Lung Cancer patients with squamous cell histology have been a traditionally hard- to-treat patient group, with less than 5% of patients with advanced SCC, surviving for five years or longer. Some of the advanced NSCLC tumors are dependent on the Epidermal Growth Factor Receptor (EGFR) for cell proliferation and survival, regardless of EGFR mutation status. TARCEVA® (Erlotinib) is a reversible EGFR Tyrosine Kinase Inhibitor and is presently approved by the FDA for the treatment of locally advanced or metastatic NSCLC, after failure of at least one prior chemotherapy regimen. GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. This kinase inhibitor is indicated for the first line treatment of patients with metastatic NSCLC, whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations.
Non Small Cell Lung Cancer patients with squamous cell histology have been a traditionally hard- to-treat patient group, with less than 5% of patients with advanced SCC, surviving for five years or longer. Some of the advanced NSCLC tumors are dependent on the Epidermal Growth Factor Receptor (EGFR) for cell proliferation and survival, regardless of EGFR mutation status. TARCEVA® (Erlotinib) is a reversible EGFR Tyrosine Kinase Inhibitor and is presently approved by the FDA for the treatment of locally advanced or metastatic NSCLC, after failure of at least one prior chemotherapy regimen. GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. This kinase inhibitor is indicated for the first line treatment of patients with metastatic NSCLC, whose tumors have Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations.
The LUX-Lung 8 is a phase III trial in which 795 patients with Stage IIIB/IV Squamous Cell Carcinoma of the lung who had progressed on first line platinum based doublet therapy, were randomized 1:1 to receive GILOTRIF 40 mg PO daily (N=398) or TARCEVA 150 mg PO daily (N=397). Treatment was given until disease progression. The median age was 65 years. Majority of the patients were male, caucasian and ex-smokers. The Primary endpoint was Progression Free Survival (PFS) and Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR), Disease Control Rate (DCR), patient reported outcomes and safety. The Primary endpoint of Progression Free Survival (PFS) was met and reported in 2014 and favored GILOTRIF® over TARCEVA®. The authors in this analysis reported the Overall Survival data, as well as updated data on Progression Free Survival and other Secondary endpoints. The median Overall Survival was 7.9 months with GILOTRIF® and 6.8 months with TARCEVA® (HR=0.81; P=0.008). This meant a 19% reduction in the risk of death with GILOTRIF® when compared to TARCEVA® and this survival advantage was consistent across all time points. The updated median Progression Free Survival for GILOTRIF® was 2.6 months vs 1.9 months for TARCEVA® (HR=0.81; P=0.01). The Disease Control Rate was 50.5% for GILOTRIF® and 39.5% with TARCEVA® (P=0.002). Based on patient reported outcomes, symptoms including cough and dyspnea were better with GILOTRIF® compared to TARCEVA®. Incidence of severe adverse events was similar with both therapies, with patients on GILOTRIF® experiencing more grade 3 diarrhea and stomatitis and patients receiving TARCEVA® experiencing more grade 3 rash. The authors concluded that GILOTRIF® should be the TKI of choice in the second line treatment of patients with Squamous Cell Carcinoma of the lung, as it significantly improves Overall Survival, Progression Free Survival, Disease Control Rate and symptom control, with manageable toxicities, when compared to TARCEVA®. Afatinib (A) vs erlotinib (E) as second-line therapy of patients (pts) with advanced squamous cell carcinoma (SCC) of the lung following platinum-based chemotherapy: Overall survival (OS) analysis from the global phase III trial LUX-Lung 8 (LL8). Soria J, Felip E, Cobo M, et al. J Clin Oncol 33, 2015 (suppl; abstr 8002)
Late Breaking Abstract - ASCO 2015: OPDIVO® Improves Overall Survival in Non- Squamous NSCLC Patients
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous cell carcinomas, 40% are Adenocarcinomas and 10% are Large cell carcinomas. The treatment paradigm for solid tumors has been rapidly evolving with a better understanding of the Immune checkpoints. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system.  Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4), also known as CD152, PD-1(Programmed cell Death-1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. The first Immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® (Ipilimumab) , an antibody that blocks Immune checkpoint protein/receptor CTLA- 4, has been shown to prolong overall survival in patients with previously treated, unresectable or metastatic melanoma. OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. The U. S. Food and Drug Administration granted approval to OPDIVO®, for the treatment of patients with metastatic Squamous Non-Small Cell Lung Cancer (NSCLC), with progression on or after platinum based chemotherapy. CheckMate 057 is a randomized, international, phase 3 study designed to evaluate the benefit of OPDIVO® for patients with Non-Squamous (NSQ) NSCLC who had progressed after platinum-based doublet chemotherapy. A total of 582 patients were randomized to receive OPDIVO® 3 mg/kg IV every 2 weeks (n=292) or TAXOTERE® 75 mg/m2 IV every 3 weeks (n=290). Eligible patients included those with advanced Non-Squamous NSCLC who had progressed after platinum-based doublet chemotherapy and a Tyrosine Kinase Inhibitor (TKI), if deemed eligible for a TKI. Treatment was continued until disease progression or unacceptable toxicity. The primary clinical endpoint was Overall Survival (OS). Secondary endpoints included Objective Response Rate (ORR), Progression Free Survival (PFS), Efficacy based on PD-L1 expression, Quality of Life, and Safety. The study was stopped earlier than expected following assessment by the independent Data Monitoring Committee (DMC) which concluded that the study met its endpoint, demonstrating superior overall survival, in patients receiving OPDIVO®, compared to the control group. Patients in the OPDIVO®, group had a significantly higher median OS compared to those in the TAXOTERE® group (12.2 months versus 9.4 months, Hazard Ratio [HR] 0.73, P=0.0015). This meant a 27% reduction in the risk of death in the OPDIVO® group and this survival benefit was seen in all predefined subgroup of patients. The Objective Response Rate (ORR) was also significantly higher for patients receiving OPDIVO® compared to TAXOTERE® (19% versus 12%, P=0.0246) and the median duration of response (DOR) was significantly higher for the OPDIVO® group (17.2 months) vs the TAXOTERE® group (5.6 months). More importantly, when tumor PD-L1 expression was correlated with Overall Survival, the median OS for OPDIVO® was 17.2 months, 18.2 months, and 19.4 months for patients with tumors having 1% or higher, 5% or higher, and 10% or higher of cells staining positive for PD-L1, respectively, compared with 9.0 months, 8.1 months, and 8.0 months with TAXOTERE® treatment. Even though this study showed significant survival outcomes for patients expressing any level of PD-L1, the magnitude of benefit was even more so, in patients with tumors expressing higher levels of PD-L1. PD-L1 expression may therefore be a predictor of response, although this should not yet be used for patient selection. Grade 3-5 adverse events occurred more often in the TAXOTERE® group compared to the OPDIVO® group (54% vs 10%). Based on this compelling data, the authors concluded that OPDIVO® significantly improves Overall Survival when compared to TAXOTERE®, in patients with advanced non-Squamous NSCLC, after failure of platinum based doublet therapy. Phase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non-squamous cell (non-SQ) non-small cell lung cancer (NSCLC). Paz-Ares L, Horn L, Borghaei H, et al. J Clin Oncol 33, 2015 (suppl; abstr LBA109)
Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4), also known as CD152, PD-1(Programmed cell Death-1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. The first Immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® (Ipilimumab) , an antibody that blocks Immune checkpoint protein/receptor CTLA- 4, has been shown to prolong overall survival in patients with previously treated, unresectable or metastatic melanoma. OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. The U. S. Food and Drug Administration granted approval to OPDIVO®, for the treatment of patients with metastatic Squamous Non-Small Cell Lung Cancer (NSCLC), with progression on or after platinum based chemotherapy. CheckMate 057 is a randomized, international, phase 3 study designed to evaluate the benefit of OPDIVO® for patients with Non-Squamous (NSQ) NSCLC who had progressed after platinum-based doublet chemotherapy. A total of 582 patients were randomized to receive OPDIVO® 3 mg/kg IV every 2 weeks (n=292) or TAXOTERE® 75 mg/m2 IV every 3 weeks (n=290). Eligible patients included those with advanced Non-Squamous NSCLC who had progressed after platinum-based doublet chemotherapy and a Tyrosine Kinase Inhibitor (TKI), if deemed eligible for a TKI. Treatment was continued until disease progression or unacceptable toxicity. The primary clinical endpoint was Overall Survival (OS). Secondary endpoints included Objective Response Rate (ORR), Progression Free Survival (PFS), Efficacy based on PD-L1 expression, Quality of Life, and Safety. The study was stopped earlier than expected following assessment by the independent Data Monitoring Committee (DMC) which concluded that the study met its endpoint, demonstrating superior overall survival, in patients receiving OPDIVO®, compared to the control group. Patients in the OPDIVO®, group had a significantly higher median OS compared to those in the TAXOTERE® group (12.2 months versus 9.4 months, Hazard Ratio [HR] 0.73, P=0.0015). This meant a 27% reduction in the risk of death in the OPDIVO® group and this survival benefit was seen in all predefined subgroup of patients. The Objective Response Rate (ORR) was also significantly higher for patients receiving OPDIVO® compared to TAXOTERE® (19% versus 12%, P=0.0246) and the median duration of response (DOR) was significantly higher for the OPDIVO® group (17.2 months) vs the TAXOTERE® group (5.6 months). More importantly, when tumor PD-L1 expression was correlated with Overall Survival, the median OS for OPDIVO® was 17.2 months, 18.2 months, and 19.4 months for patients with tumors having 1% or higher, 5% or higher, and 10% or higher of cells staining positive for PD-L1, respectively, compared with 9.0 months, 8.1 months, and 8.0 months with TAXOTERE® treatment. Even though this study showed significant survival outcomes for patients expressing any level of PD-L1, the magnitude of benefit was even more so, in patients with tumors expressing higher levels of PD-L1. PD-L1 expression may therefore be a predictor of response, although this should not yet be used for patient selection. Grade 3-5 adverse events occurred more often in the TAXOTERE® group compared to the OPDIVO® group (54% vs 10%). Based on this compelling data, the authors concluded that OPDIVO® significantly improves Overall Survival when compared to TAXOTERE®, in patients with advanced non-Squamous NSCLC, after failure of platinum based doublet therapy. Phase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non-squamous cell (non-SQ) non-small cell lung cancer (NSCLC). Paz-Ares L, Horn L, Borghaei H, et al. J Clin Oncol 33, 2015 (suppl; abstr LBA109)
Targeting ROS1 Molecular Driver Mutations with XALKORI® in NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous cell carcinomas, 40% are Adenocarcinomas and 10% are Large cell carcinomas. There is now growing body of evidence suggesting superior outcomes when advanced NSCLC patients with specific genomic alterations receive targeted therapies.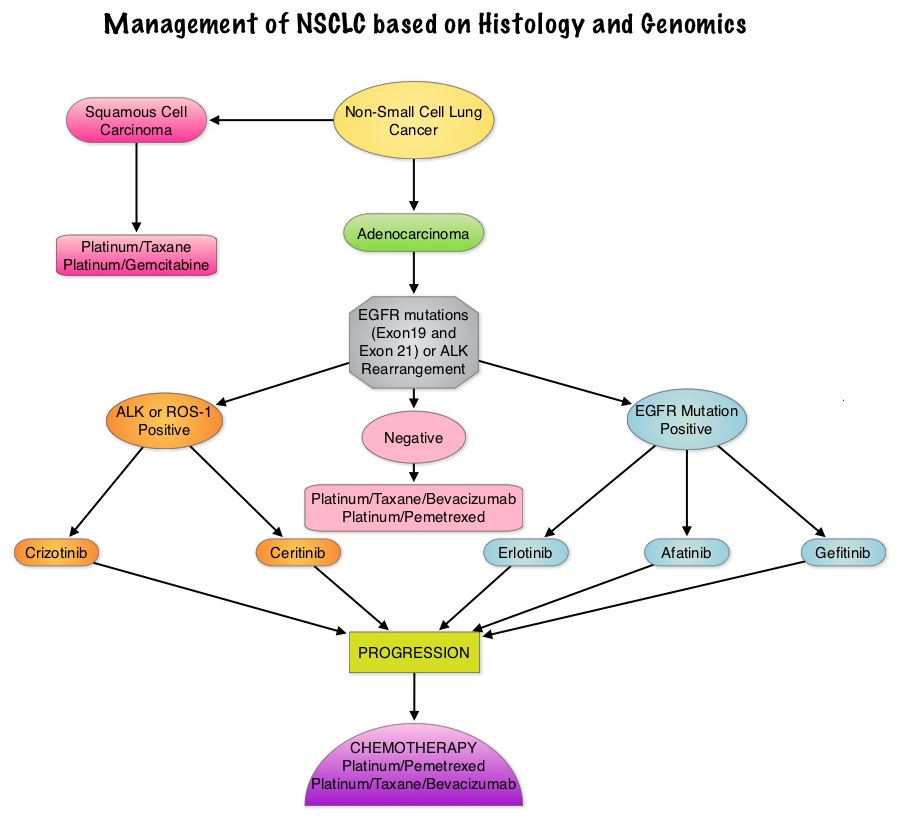 Approximately 1% - 2% of lung adenocarcinomas harbor ROS1 gene rearrangements. ROS1 gene is located on chromosome 6q22 (long arm of chromosome 6) and plays an important role in cell growth and development. ROS1 gene fusion with another gene results in a mutated DNA sequence which then produces an abnormal protein responsible for unregulated cell growth and cancer. ROS1 gene rearrangement has been identified as a driver mutation in Non Small cell Lung Cancer with adenocarcinoma histology. This is more common in nonsmokers or in light smokers (<10 pack years) who are relatively young (average age of 50 years) and thus share similar characteristics with ALK-positive patients. The ROS protein and the ALK protein have similar structure and function and are sensitive to Tyrosine Kinase Inhibitors such as XALKORI® (Crizotinib) and ZYKADIA® (Ceritinib). ROS1 mutations have been also been associated with Cholangiocarcinoma (Bile duct cancer) and Glioblastoma multiforme. ROS1 rearrangements are mutually exclusive with other oncogenic mutations found in NSCLC such as EGFR mutations, KRAS mutations and ALK rearrangement. The presence of a ROS1 rearrangement can be detected by Fluorescence In Situ Hybridization (FISH), ImmunoHistoChemistry (IHC), Reverse Transcriptase– Polymerase Chain Reaction (RT-PCR) and Next Generation-Sequencing. XALKORI® is a small molecule Tyrosine Kinase Inhibitor that targets ALK, MET and ROS1 tyrosine kinases. In a previously published expansion cohort of the phase 1 study by Shaw and colleagues ( NEJM 2014; 371:1963-1971), XALKORI® showed significant activity in patients with in patients with advanced ROS1rearranged NSCLC. The authors in this publication provided additional evidence that ROS1 gene rearrangement is an actionable target in NSCLC, by conducting a retrospective study in centers that tested for ROS1 rearrangement and evaluated the outcomes of ROS1-positive NSCLC patients, who had been treated with XALKORI®. They included 32 patients with NSCLC whose tumors showed ROS1 rearrangement and who had received off-label treatment with XALKORI®. The median age was 50.5 years. They noted an overall response rate of 80% and a disease control rate, 86.7%. The median Progression Free Survival (PFS) was 9.1 months, and the PFS rate at 12 months was 44%. This impressive efficacy data again validates that similar to EGFR mutations and ALK rearrangements, ROS1 gene rearrangements are molecular drivers and patients with NSCLC with adenocarcinoma histology should be tested for ROS1. Crizotinib Therapy for Advanced Lung Adenocarcinoma and a ROS1 Rearrangement: Results From the EUROS1 Cohort. Mazières J, Zalcman G, Crinò L, J Clin Oncol. 2015;33:992-999
Approximately 1% - 2% of lung adenocarcinomas harbor ROS1 gene rearrangements. ROS1 gene is located on chromosome 6q22 (long arm of chromosome 6) and plays an important role in cell growth and development. ROS1 gene fusion with another gene results in a mutated DNA sequence which then produces an abnormal protein responsible for unregulated cell growth and cancer. ROS1 gene rearrangement has been identified as a driver mutation in Non Small cell Lung Cancer with adenocarcinoma histology. This is more common in nonsmokers or in light smokers (<10 pack years) who are relatively young (average age of 50 years) and thus share similar characteristics with ALK-positive patients. The ROS protein and the ALK protein have similar structure and function and are sensitive to Tyrosine Kinase Inhibitors such as XALKORI® (Crizotinib) and ZYKADIA® (Ceritinib). ROS1 mutations have been also been associated with Cholangiocarcinoma (Bile duct cancer) and Glioblastoma multiforme. ROS1 rearrangements are mutually exclusive with other oncogenic mutations found in NSCLC such as EGFR mutations, KRAS mutations and ALK rearrangement. The presence of a ROS1 rearrangement can be detected by Fluorescence In Situ Hybridization (FISH), ImmunoHistoChemistry (IHC), Reverse Transcriptase– Polymerase Chain Reaction (RT-PCR) and Next Generation-Sequencing. XALKORI® is a small molecule Tyrosine Kinase Inhibitor that targets ALK, MET and ROS1 tyrosine kinases. In a previously published expansion cohort of the phase 1 study by Shaw and colleagues ( NEJM 2014; 371:1963-1971), XALKORI® showed significant activity in patients with in patients with advanced ROS1rearranged NSCLC. The authors in this publication provided additional evidence that ROS1 gene rearrangement is an actionable target in NSCLC, by conducting a retrospective study in centers that tested for ROS1 rearrangement and evaluated the outcomes of ROS1-positive NSCLC patients, who had been treated with XALKORI®. They included 32 patients with NSCLC whose tumors showed ROS1 rearrangement and who had received off-label treatment with XALKORI®. The median age was 50.5 years. They noted an overall response rate of 80% and a disease control rate, 86.7%. The median Progression Free Survival (PFS) was 9.1 months, and the PFS rate at 12 months was 44%. This impressive efficacy data again validates that similar to EGFR mutations and ALK rearrangements, ROS1 gene rearrangements are molecular drivers and patients with NSCLC with adenocarcinoma histology should be tested for ROS1. Crizotinib Therapy for Advanced Lung Adenocarcinoma and a ROS1 Rearrangement: Results From the EUROS1 Cohort. Mazières J, Zalcman G, Crinò L, J Clin Oncol. 2015;33:992-999
Post Operative Radiation Therapy (PORT) Improves Survival in Resected N2 Non-Small Cell Lung Cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and over 158,000 patients will die of the disease. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Based on the extent of the disease and the treatment approach, patients with localized or locally advanced NSCLC can be divided into two groups – 1) Surgically resectable disease group (stage I, stage II, and selected stage III tumors) for whom postoperative Cisplatin-based combination chemotherapy may provide a survival advantage (particularly for those with resected stage II or stage IIIA NSCLC). 2) Locally (T3–T4) and/or regionally (N2–N3) advanced disease group who have unresectable disease, who benefit with radiation therapy in combination with chemotherapy. Based on available evidence, postoperative chemotherapy is not recommended outside of a clinical trial for patients with completely resected stage I NSCLC.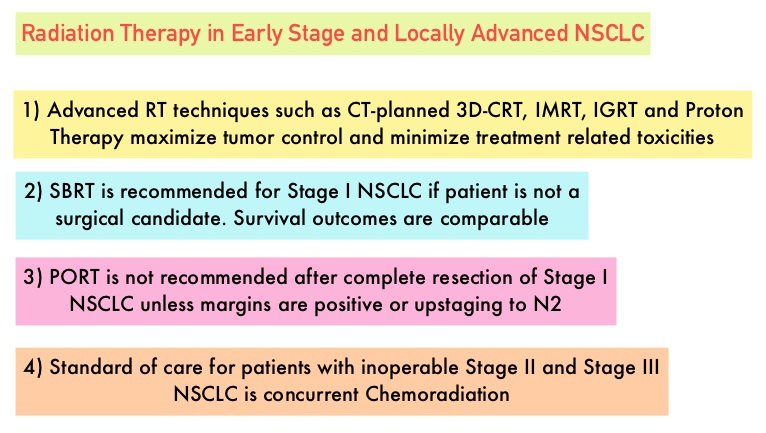 Although there is sufficient evidence for postoperative chemotherapy in patients with stage II or stage IIIA NSCLC, its usefulness in patients with stage IB NSCLC remains unclear. The value of adjuvant Post Operative Radiation Therapy (PORT) has been evaluated and has not been found to improve the outcome of patients with completely resected stage I NSCLC. The risk of locoregional recurrence (LRR) is 20-40% in patients with resected node-positive disease and this in turn may independently contribute to worsened Overall Survival in patients with NSCLC. Nonetheless, a large meta-analysis from trials conducted mainly in the 1960’s and 1970’s showed that adjuvant Post Operative Radiation Therapy (PORT) in stages IIA NSCLC and IIB NSCLC was associated with an 18% relative increase in the risk of death compared with surgery alone. This decrease in Overall Survival has been attributed to outmoded RT techniques and doses resulting in cardiac and pulmonary toxicity. This study was conducted to evaluate the impact of modern (Computed Tomography simulation and at least Linear accelerator- Linac based, three-dimensional, conformal RT) Post Operative Radiation Therapy (PORT) on Overall Survival (OS) in a large population-based registry of patients with completely resected stage IIIA (N2) NSCLC, when compared with adjuvant chemotherapy alone. The authors identified 4,483 patients in the National Cancer Data Base from 2006 to 2010 with pathologic N2, NSCLC, who underwent complete resection and adjuvant chemotherapy. This large patient population was representative of typical patients treated throughout the United States. Of these large cohort of patients, 1,850 had received PORT (45 Gy or more) and 2,633 patients did not. The authors evaluated the impact of patient and treatment variables on OS. The median follow-up time was 22 months. The use of PORT was associated with an increase in median and 5-year OS compared with no PORT (median OS, 45.2 vs 40.7 months, respectively; 5-year OS, 39.3% vs 34.8% respectively; P= 0.014). The improved OS remained, independently predicted by younger age, female sex, urban population, fewer comorbidities, smaller tumor size, multiagent chemotherapy, resection with at least a lobectomy, and PORT. The authors concluded that modern Post Operative Radiotherapy Therapy confers an additional OS advantage beyond that achieved with adjuvant chemotherapy alone, for patients with N2, NSCLC after complete resection and adjuvant chemotherapy. Postoperative Radiotherapy for Pathologic N2 Non–Small-Cell Lung Cancer Treated With Adjuvant Chemotherapy: A Review of the National Cancer Data Base. Robinson CG, Patel AP, Bradley JD, et al. J Clin Oncol. 2015; 33:870-876
Although there is sufficient evidence for postoperative chemotherapy in patients with stage II or stage IIIA NSCLC, its usefulness in patients with stage IB NSCLC remains unclear. The value of adjuvant Post Operative Radiation Therapy (PORT) has been evaluated and has not been found to improve the outcome of patients with completely resected stage I NSCLC. The risk of locoregional recurrence (LRR) is 20-40% in patients with resected node-positive disease and this in turn may independently contribute to worsened Overall Survival in patients with NSCLC. Nonetheless, a large meta-analysis from trials conducted mainly in the 1960’s and 1970’s showed that adjuvant Post Operative Radiation Therapy (PORT) in stages IIA NSCLC and IIB NSCLC was associated with an 18% relative increase in the risk of death compared with surgery alone. This decrease in Overall Survival has been attributed to outmoded RT techniques and doses resulting in cardiac and pulmonary toxicity. This study was conducted to evaluate the impact of modern (Computed Tomography simulation and at least Linear accelerator- Linac based, three-dimensional, conformal RT) Post Operative Radiation Therapy (PORT) on Overall Survival (OS) in a large population-based registry of patients with completely resected stage IIIA (N2) NSCLC, when compared with adjuvant chemotherapy alone. The authors identified 4,483 patients in the National Cancer Data Base from 2006 to 2010 with pathologic N2, NSCLC, who underwent complete resection and adjuvant chemotherapy. This large patient population was representative of typical patients treated throughout the United States. Of these large cohort of patients, 1,850 had received PORT (45 Gy or more) and 2,633 patients did not. The authors evaluated the impact of patient and treatment variables on OS. The median follow-up time was 22 months. The use of PORT was associated with an increase in median and 5-year OS compared with no PORT (median OS, 45.2 vs 40.7 months, respectively; 5-year OS, 39.3% vs 34.8% respectively; P= 0.014). The improved OS remained, independently predicted by younger age, female sex, urban population, fewer comorbidities, smaller tumor size, multiagent chemotherapy, resection with at least a lobectomy, and PORT. The authors concluded that modern Post Operative Radiotherapy Therapy confers an additional OS advantage beyond that achieved with adjuvant chemotherapy alone, for patients with N2, NSCLC after complete resection and adjuvant chemotherapy. Postoperative Radiotherapy for Pathologic N2 Non–Small-Cell Lung Cancer Treated With Adjuvant Chemotherapy: A Review of the National Cancer Data Base. Robinson CG, Patel AP, Bradley JD, et al. J Clin Oncol. 2015; 33:870-876
OPDIVO® now Approved for Advanced, Refractory Squamous Non-Small Cell Lung Cancer
SUMMARY: The U. S. Food and Drug Administration granted approval to Nivolumab (OPDIVO®) for the treatment of patients with metastatic squamous Non-Small Cell Lung Cancer (NSCLC) with progression on or after platinum based chemotherapy. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 221,200 new cases of lung cancer will be diagnosed in the United States in 2015 and a over 158,000 patients will die of the disease. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous cell carcinomas, 40% are Adenocarcinomas and 10% are Large cell carcinomas. Non-Small Cell Lung Cancer patients with Squamous cell histology have been a traditionally hard- to-treat patient group. 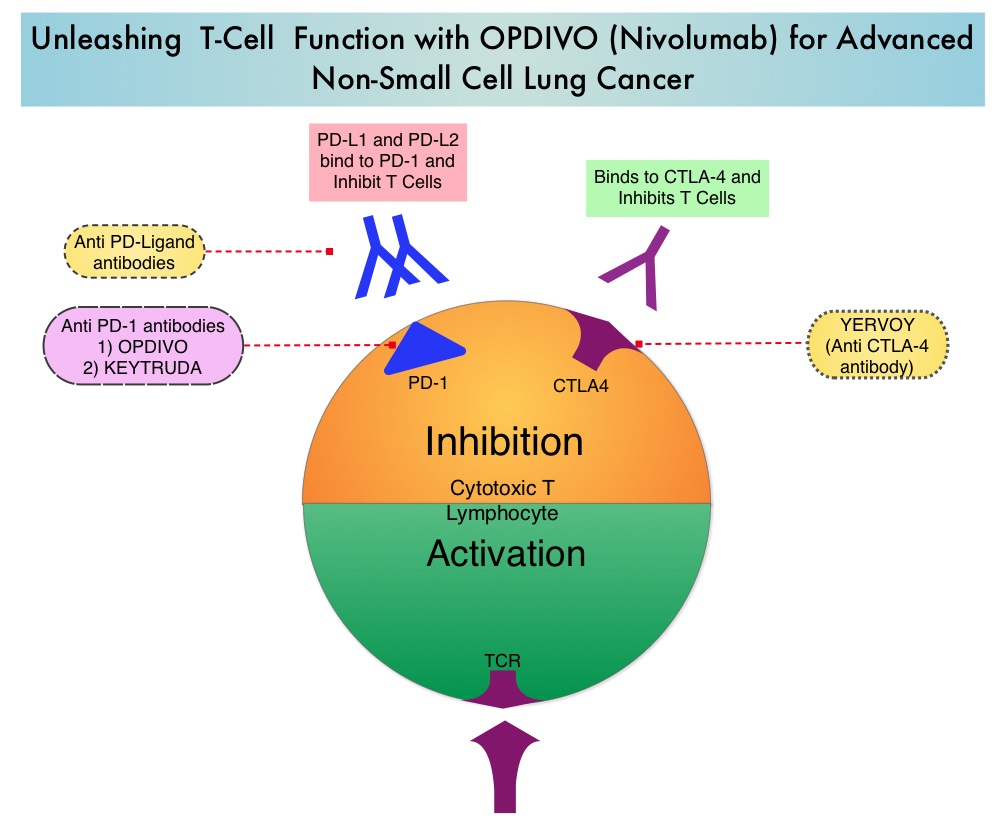 The development a novel immunotherapeutic approaches, with a better understanding of the Immune checkpoints has however changed the treatment paradigm. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4), also known as CD152, PD-1(Programmed cell Death-1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. The first Immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® (Ipilimumab), an antibody that blocks Immune checkpoint protein/receptor CTLA- 4, has been shown to prolong overall survival in patients with previously treated, unresectable or metastatic melanoma. OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. The approval of OPDIVO® was based on CheckMate-017, an open label, multicenter, multinational randomized phase III trial in which 272 patients with metastatic squamous NSCLC who had experienced disease progression during or after one prior platinum-based chemotherapy regimen were randomized to receive OPDIVO® (Nivolumab) 3 mg/kg IV every 2 weeks (N=135) or TAXOTERE® (Docetaxel) 75 mg/m2 IV every 3 weeks (N=137). The primary endpoint was Overall Survival (OS) and secondary endpoints included Progression Free Survival (PFS) and Objective Response Rate (ORR). This study was stopped early at the protocol pre-specified interim analysis after an independent monitoring panel determined that the primary endpoint of improved Overall Survival (OS) with OPDIVO® had been reached. The median OS was 9.2 months for patients assigned to OPDIVO® and 6 months for those in the TAXOTERE® group (HR=0.59; P=0.00025). This suggested a 41% improvement in the OS with OPDIVO® compared to TAXOTERE®. This FDA approval was further supported by a single arm, multinational, multicenter trial in patients with metastatic squamous NSCLC (N=117) who had progressed after receiving a platinum-based therapy and at least one additional systemic regimen. OPDIVO® in this study, was administered as a single agent at 3mg/kg IV every two weeks until disease progression or treatment discontinuation. The primary endpoint was Objective Response Rate (ORR) and exploratory endpoints were Overall Survival (OS), Progression Free Survival (PFS) and efficacy, based on PD-L1 expression status. With 11 months of minimum follow up, the Objective Response Rate (ORR) was 15% independent of PD-L1 status. The estimated one-year survival rate was 41% and median Overall Survival was 8.2 months. The authors noted that an additional 26% of patients had stable disease for a median duration of 6 months, resulting in a disease control rate (ORR+stable disease) of 41%. The most frequent grade 3-4 adverse events noted in at least 5% of the patients were fatigue, dyspnea and musculoskeletal pain. OPDIVO® will now be a new treatment option, with survival advantage, for patients with advanced relapsed and refractory metastatic squamous NSCLC. Phase II study of nivolumab (Anti-PD-1, BMS-936558, ONO-4538) in patients with advanced, refractory squamous non-small cell lung cancer. Ramalingam SS, Mazieres J, Planchard D, et al. Presented at: 2014 Multidisciplinary Symposium in Thoracic Oncology; Chicago, IL. LBA#3462
The development a novel immunotherapeutic approaches, with a better understanding of the Immune checkpoints has however changed the treatment paradigm. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4), also known as CD152, PD-1(Programmed cell Death-1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. The first Immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® (Ipilimumab), an antibody that blocks Immune checkpoint protein/receptor CTLA- 4, has been shown to prolong overall survival in patients with previously treated, unresectable or metastatic melanoma. OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. The approval of OPDIVO® was based on CheckMate-017, an open label, multicenter, multinational randomized phase III trial in which 272 patients with metastatic squamous NSCLC who had experienced disease progression during or after one prior platinum-based chemotherapy regimen were randomized to receive OPDIVO® (Nivolumab) 3 mg/kg IV every 2 weeks (N=135) or TAXOTERE® (Docetaxel) 75 mg/m2 IV every 3 weeks (N=137). The primary endpoint was Overall Survival (OS) and secondary endpoints included Progression Free Survival (PFS) and Objective Response Rate (ORR). This study was stopped early at the protocol pre-specified interim analysis after an independent monitoring panel determined that the primary endpoint of improved Overall Survival (OS) with OPDIVO® had been reached. The median OS was 9.2 months for patients assigned to OPDIVO® and 6 months for those in the TAXOTERE® group (HR=0.59; P=0.00025). This suggested a 41% improvement in the OS with OPDIVO® compared to TAXOTERE®. This FDA approval was further supported by a single arm, multinational, multicenter trial in patients with metastatic squamous NSCLC (N=117) who had progressed after receiving a platinum-based therapy and at least one additional systemic regimen. OPDIVO® in this study, was administered as a single agent at 3mg/kg IV every two weeks until disease progression or treatment discontinuation. The primary endpoint was Objective Response Rate (ORR) and exploratory endpoints were Overall Survival (OS), Progression Free Survival (PFS) and efficacy, based on PD-L1 expression status. With 11 months of minimum follow up, the Objective Response Rate (ORR) was 15% independent of PD-L1 status. The estimated one-year survival rate was 41% and median Overall Survival was 8.2 months. The authors noted that an additional 26% of patients had stable disease for a median duration of 6 months, resulting in a disease control rate (ORR+stable disease) of 41%. The most frequent grade 3-4 adverse events noted in at least 5% of the patients were fatigue, dyspnea and musculoskeletal pain. OPDIVO® will now be a new treatment option, with survival advantage, for patients with advanced relapsed and refractory metastatic squamous NSCLC. Phase II study of nivolumab (Anti-PD-1, BMS-936558, ONO-4538) in patients with advanced, refractory squamous non-small cell lung cancer. Ramalingam SS, Mazieres J, Planchard D, et al. Presented at: 2014 Multidisciplinary Symposium in Thoracic Oncology; Chicago, IL. LBA#3462
Molecular Testing for Selection of Patients with Lung Cancer for Epidermal Growth Factor Receptor and Anaplastic Lymphoma Kinase Tyrosine Kinase Inhibitors: American Society of Clinical Oncology Endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Guideline
SUMMARY: There is now growing body of evidence suggesting superior outcomes when advanced NSCLC patients with specific genomic alterations receive targeted therapies. Following review of 127 studies by experts and input from a scientific advisory panel, The College of American Pathologists (CAP), the International Association for the Study of Lung Cancer (IASLC), and the Association for Molecular Pathology (AMP) offered evidence-based recommendations for the molecular analysis of lung cancers for Epidermal Growth Factor Receptor (EGFR ) mutations and Anaplastic Lymphoma Kinase (ALK) rearrangements, thereby selecting patients with lung cancer, for treatment with EGFR and ALK tyrosine kinase inhibitors. The ASCO review panel has endorsed these guidelines which specifically address the following questions:
1) Which patients should be tested for EGFR mutations and ALK rearrangements?
EGFR or ALK testing is recommended for all patients with advanced lung adenocarcinoma or tumors with an adenocarcinoma component, irrespective of clinical characteristics such as smoking history, sex, race, or other clinical factors. Tumor samples of other histologies for which an adenocarcinoma component cannot be excluded because of sampling, can be considered for testing, particularly if clinical criteria are suggestive (eg, younger age, lack of smoking history). Both primary tumors and metastatic lesions are suitable for testing. When fully excised lung cancer specimens are available, EGFR and ALK testing is not recommended in lung cancers that lack any adenocarcinoma component, such as pure squamous cell carcinomas, pure small-cell carcinomas, or large-cell carcinomas lacking IHC (ImmunoHistoChemistry) evidence of adenocarcinoma differentiation.
2) When should a patient specimen be tested for EGFR mutation or ALK rearrangement?
Testing should be ordered at the time of diagnosis of advanced disease or recurrence. For patients with earlier stage disease who undergo surgical resection, testing at the time of diagnosis is encouraged so that molecular information is available to an oncologist at the time of recurrence, for a subset of patients who subsequently experience recurrence. Tissue should be prioritized for EGFR and ALK testing.
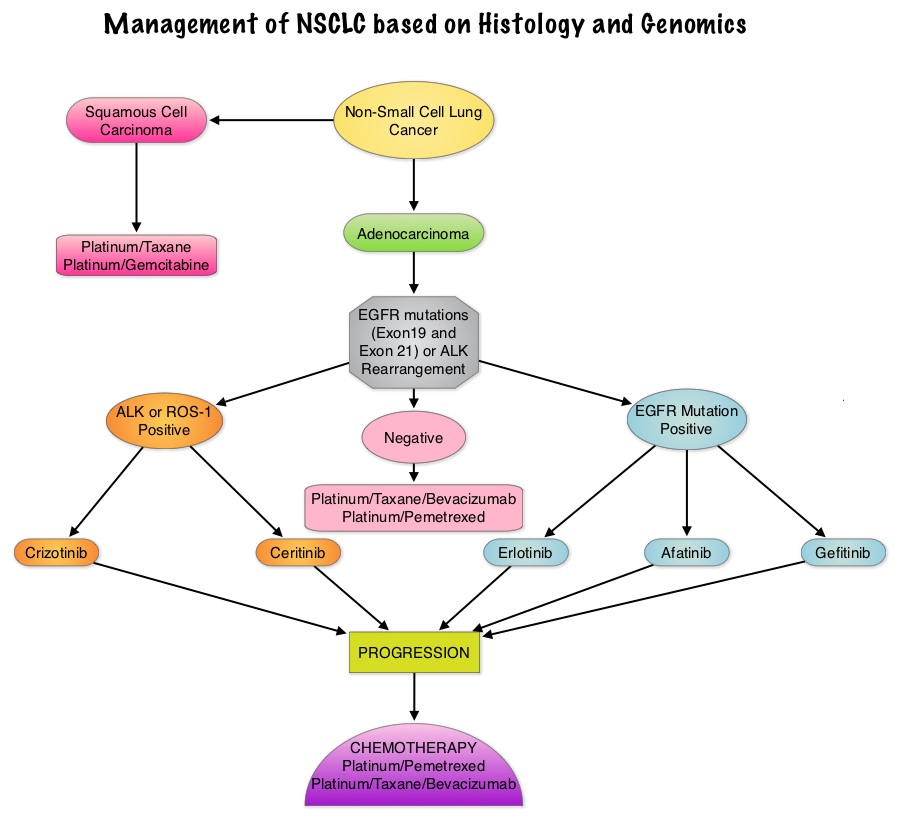 3) How rapidly should test results be available?
3) How rapidly should test results be available?
Laboratory turnaround times of 5 to 10 working days (2 weeks) for EGFR and ALK results are recommended.
4) How should specimens be processed for EGFR mutation testing?
Pathologists should use Formalin-Fixed, Paraffin-Embedded (FFPE) specimens or fresh frozen or alcohol-fixed specimens for PCR based EGFR mutation tests. EGFR and ALK testing can be performed with cytology samples, with cell blocks being preferred over smear preparations.
5) How should EGFR testing be performed?
EGFR testing should detect mutations in samples composed of as few as 50% tumor cells, although sensitivity to detect mutations in samples containing > 10% tumor cells is strongly encouraged. Sensitizing EGFR mutations with a population frequency of at least 1% should be reported. IHC for total EGFR as well as EGFR copy number analysis by FISH (Fluorescence In Situ Hybridization) is not recommended.
6) What is the role of KRAS analysis in selecting patients for targeted therapy with EGFR TKIs?
KRAS mutations are common (30%) in lung adenocarcinomas and mutually exclusive with EGFR and ALK. Testing for KRAS may be performed initially to exclude KRAS mutated tumors from EGFR and ALK testing but KRAS mutation testing is not recommended as a sole determinant of EGFR-targeted therapy.
7) What additional testing considerations are important in the setting of secondary or acquired EGFR TKI resistance?
If a laboratory performs testing on specimens from patients with acquired resistance to EGFR kinase inhibitors, such tests should be able to detect the secondary EGFR T790M mutation in as few as 5% of cells.
8) What methods should be used for ALK testing?
ALK FISH assay using dual labeled break-apart probes should be used for selecting patients for ALK TKI therapy. ALK IHC, if carefully validated, may be considered as a screening methodology to select specimens for ALK FISH testing. RT-PCR (Reverse Transcription–Polymerase Chain Reaction) is not recommended as an alternative to FISH, for selecting patients for ALK inhibitor therapy.
9) Are other molecular markers suitable for testing in lung cancer?
Testing for EGFR should be prioritized over other molecular markers in lung adenocarcinoma followed by testing for ALK. Testing for ROS1 and RET rearrangements may soon become a part of the guidelines.
10) How should molecular testing of lung adenocarcinomas be implemented and operationalized?
Pathology departments should establish a process wherein tissue (blocks or unstained slides) is sent to outside molecular laboratories within 3 days of receiving a request and to in house molecular laboratories within 24 hours. Results should be available within 2 weeks and reported in a format that is easily understood by oncologists and nonspecialist pathologists.
Leighl NB, Rekhtman N, Biermann WA, et al. J Clin Oncol 2014;32:3673-3679
Screening for Lung Cancer: U.S. Preventive Services Task Force Recommendation Statement
SUMMARY: The Centers for Medicare & Medicaid Services (CMS) on November 14, 2014, proposed that the evidence is sufficient, to add a Lung cancer screening counseling and shared decision making visit for appropriate beneficiaries. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 224,000 new cases of lung cancer will be diagnosed in the United States in 2014 and over 159,000 will die of the disease. Given the incidence and mortality related to Lung cancer, several studies were conducted dating back to the 1960’s and 1970’s in an attempt to find an appropriate screening test for Lung cancer. They included testing sputum cytology and chest radiography or a combination of both. However, these screening methodologies did not conclusively demonstrate improvements in health outcomes. The results of a NCI-sponsored National Lung Screening Trial (NLST) published in 2011, was more optimistic. In this federally funded U.S. study, 53,439 asymptomatic participants, 55 to 74 years of age, with at least 30 pack-year smoking history were enrolled and randomized to undergo annual screening with either Low dose CT scan (n=26,715) or a chest X-Ray (n=26,724), for three years. The use of Low Dose CT (LDCT) scans for 3 years in this high risk, healthy patients, resulted in a 20% reduction in Lung cancer mortality, compared to screening with a chest X-Ray. Based on these findings, Lung cancer screening was felt appropriate for the following groups of patients:
1) People 55-74 years of age with no signs or symptoms of Lung disease or lung Cancer
2) Current or former smoker with a 30 pack year smoking history (Number of years smoked multiplied by the number of packs of cigarettes per day with each pack containing 20 cigarettes)
3) Former smokers who has quit smoking within the past 15 years
The United States Preventive Services Task Force (USPSTF) recommended annual screening for lung cancer with Low Dose Computed Tomography in adult individuals, between ages 55 to 80 years who have a 30 pack-year smoking history and currently smoke or have quit within the past 15 years.  Screening should be discontinued once a person has not smoked for 15 years or develops a health problem that substantially limits life expectancy or the ability or willingness to have curative lung surgery. This was a Grade: B recommendation which meant that the USPSTF recommends the service and there is high certainty that the net benefit is moderate or there is moderate certainty that the net benefit is moderate to substantial. This therefore meant that clinicians offer or provide this service to these high risk individuals.
Screening should be discontinued once a person has not smoked for 15 years or develops a health problem that substantially limits life expectancy or the ability or willingness to have curative lung surgery. This was a Grade: B recommendation which meant that the USPSTF recommends the service and there is high certainty that the net benefit is moderate or there is moderate certainty that the net benefit is moderate to substantial. This therefore meant that clinicians offer or provide this service to these high risk individuals.
Based on this information the Centers for Medicare & Medicaid Services (CMS) on November 14, 2014, proposed that the evidence is sufficient, to add a Lung cancer screening counseling and shared decision making visit. CMS proposed, screening for Lung cancer with Low Dose Computed Tomography (LDCT), for appropriate beneficiaries, once per year, as an additional preventive service benefit under the Medicare program, only if all of the following criteria are met:
1. Age 55-74 years
2. Asymptomatic (no signs or symptoms of lung disease)
3. Tobacco smoking history of at least 30 pack-years (one pack-year = smoking one pack per day for one year; 1 pack = 20 cigarettes)
4. Current smoker or one who has quit smoking within the last 15 years
5. A lung cancer screening counseling and shared decision making visit which includes the use of one or more decision aids discussing the benefits, harms, follow-up diagnostic testing, over-diagnosis, false positive rate, and total radiation exposure
6. Counseling on the importance of adherence to annual LDCT lung cancer screening, impact of comorbidities and ability or willingness to undergo diagnosis and treatment
7. Counseling on the importance of maintaining cigarette smoking abstinence if former smoker, or smoking cessation if current smoker and, if appropriate, offering additional Medicare-covered tobacco cessation counseling services
Lung Cancer screening is performed using a non-contrast, Low Dose CT scan (LDCT) at an accredited advanced diagnostic imaging center with an effective radiation dose less than 1.5 mSv (the equivalent of 15 chest x-rays), compared to a standard chest CT with a median radiation dose of 8 mSv. The imaging center must collect and submit required data to a CMS-approved national registry for each LDCT lung cancer screening performed. Moyer VA, et al. on behalf of the U.S. Preventive Services Task Force. Ann Intern Med. 2014;160:330-338.
Phase II study of nivolumab (Anti-PD-1, BMS-936558, ONO-4538) in patients with advanced, refractory squamous non-small cell lung cancer
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 224,000 new cases of lung cancer will be diagnosed in the United States in 2014 and over 159,000 will die of the disease. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous cell carcinomas, 40% are Adenocarcinomas and 10% are Large cell carcinomas. With a better understanding of the Immune checkpoints, the gates are now wide open for the development of various immunotherapies. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent related to their ability to escape immune surveillance by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. Checkmate -063 is a Phase II single arm, open label study designed to evaluate the efficacy of OPDIVO® (Nivolumab) in patients with advanced NSCLC with squamous histology, who had progressed on platinum based therapy as well as at least one additional systemic therapy. OPDIVO® is an immune checkpoint PD-1 (Programmed cell Death 1) targeted, fully human, immunoglobulin G4 monoclonal antibody, which demonstrated an objective response in 20% - 25% of patients with advanced Non Small Cell Lung Cancer, Melanoma and Renal Cell Carcinoma, with favorable toxicities, in previously published studies. This study enrolled 117 patients and two thirds of the patients had previously failed 3 or more treatments and three fourths of patients were within 3 months of completion of their most recent therapy. OPDIVO® was administered as a single agent at 3mg/kg by intravenous infusion every two weeks until disease progression or treatment discontinuation. The primary endpoint was Objective Response Rate (ORR) and exploratory endpoints were overall survival (OS), Progression Free Survival (PFS) and efficacy, based on PD-L1 expression status. With 11 months of minimum follow up, the Objective Response Rate (ORR) was 15% as assessed by an independent review committee and the median duration of response was not reached. These responses were independent of PD-L1 status for patients with quantifiable PD-L1 expression. The estimated one-year survival rate was 41% and median Overall Survival was 8.2 months. The authors noted that an additional 26% of patients had stable disease for a median duration of 6 months, resulting in a disease control rate (ORR+stable disease) of 41%. Approximately 17% of the patients experienced grade 3-4 adverse events which included fatigue, pneumonitis and diarrhea. The authors concluded that the high response rates, median duration of response and disease control rates for Squamous NSCLC, is very promising in this difficult to treat group of patients and phase III trials are underway evaluating OPDIVO® monotherapy in frontline and previously treated patients with Non Small Cell Lung cancer. Ramalingam SS, Mazieres J, Planchard D, et al. Presented at: 2014 Multidisciplinary Symposium in Thoracic Oncology; October 30-November 1, 2014; Chicago, IL. LBA#3462
Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors vs Conventional Chemotherapy in Non–Small Cell Lung Cancer Harboring Wild-Type Epidermal Growth Factor Receptor - A Meta-analysis
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 224,000 new cases of lung cancer will be diagnosed in the United States in 2014 and over 159,000 will die of the disease. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, adenocarcinoma now is the most frequent histologic subtype of lung cancer.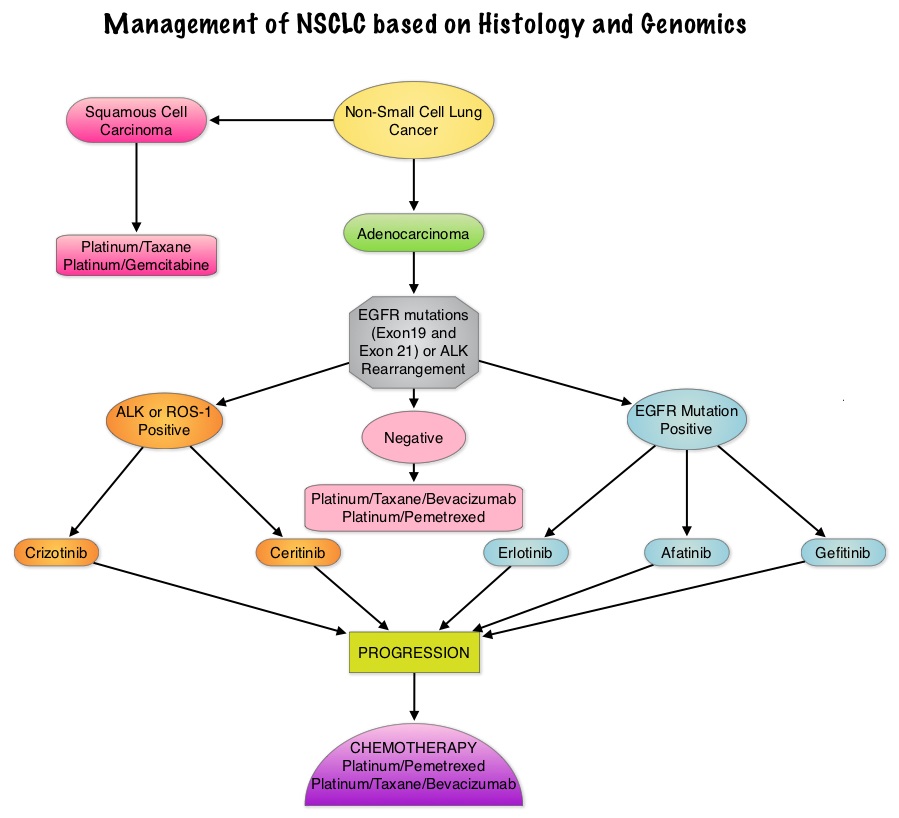 In 2004, the discovery of Epidermal Growth Factor Receptor (EGFR) mutations in some advanced Non Small Cell Lung Cancer (NSCLC) cases with adenocarcinoma histology and the favorable responses with EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib) and IRESSA® (Gefitinib), changed the treatment paradigm in favor of targeted therapy, for this patient subset. It is estimated that approximately 10% of Western patient population and 50% of Asian patients with NSCLC, harbor EGFR activating mutations. EGFR Tyrosine Kinase Inhibitors have been shown to be superior to conventional chemotherapy in this patient group with improved Progression Free Survival (PFS) and Objective Response Rates. Patients with NSCLC should therefore be tested for the most common sensitizing mutations such as deletions in exon 19 and L858R point mutations in exon 21, as these patients clearly benefit from first line EGFR TKIs. EGFR expression by IHC (ImmunoHistoChemical) staining, EGFR gene copy by FISH (Fluorescence In Situ Hybridization) and blood based proteonomic testing by VERISTRAT® is currently not recommended for the selection of first line EGFR TKIs. There is presently no evidence indicating superiority of TKIs when compared with conventional chemotherapy for the second or third line treatment of EGFR Wild Type NSCLC. Nonetheless, TKIs are often recommended due to their acceptable toxicities. To address this treatment dilemma, the authors performed a systematic review and meta-analysis of randomized controlled trials, comparing first-generation EGFR TKIs (TARCEVA® and IRESSA®) treatment with conventional chemotherapy, in patients with advanced NSCLC, harboring Wild Type EGFR. This pooled analysis included 1605 patients from 11 clinical trials, with EGFR Wild Type NSCLC. The primary outcome measured was Progression Free Survival (PFS) and secondary outcomes were Objective Response Rate and Overall Survival. It was noted in this analysis that conventional chemotherapy was associated with longer PFS, compared with EGFR TKIs, among patients harboring Wild Type EGFR tumors. The authors noted that there was significant PFS benefit with chemotherapy, in trials using more sensitive EGFR mutation analysis platforms, than direct Sanger sequencing, and this may be the result of identifying the “true” Wild Type EGFR tumors. The objective response rate was higher at 16.8% with chemotherapy versus 7.2% for TKIs. There was however no statistically significant difference in the overall survival between the chemotherapy and TKI groups. When subgroups of patients in these trials were analyzed, outcomes were similar regardless of line of treatment, dominant ethinicity or EGFR mutation analysis method. The lack of improvement in Overall Survival in the chemotherapy group has been attributed to the large cross over rates in the trials that were analyzed. The authors concluded that conventional chemotherapy is associated with superior Progression Free Survival and Objective Response Rates, in patients with advanced NSCLC, harboring Wild Type EGFR tumors, compared with EGFR TKIs and the present guidelines recommending EGFR TKIs in this patient group has to be reevaluated. Lee J, Hahn S, Kim D, et al. JAMA 2014;311:1430-1437
In 2004, the discovery of Epidermal Growth Factor Receptor (EGFR) mutations in some advanced Non Small Cell Lung Cancer (NSCLC) cases with adenocarcinoma histology and the favorable responses with EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib) and IRESSA® (Gefitinib), changed the treatment paradigm in favor of targeted therapy, for this patient subset. It is estimated that approximately 10% of Western patient population and 50% of Asian patients with NSCLC, harbor EGFR activating mutations. EGFR Tyrosine Kinase Inhibitors have been shown to be superior to conventional chemotherapy in this patient group with improved Progression Free Survival (PFS) and Objective Response Rates. Patients with NSCLC should therefore be tested for the most common sensitizing mutations such as deletions in exon 19 and L858R point mutations in exon 21, as these patients clearly benefit from first line EGFR TKIs. EGFR expression by IHC (ImmunoHistoChemical) staining, EGFR gene copy by FISH (Fluorescence In Situ Hybridization) and blood based proteonomic testing by VERISTRAT® is currently not recommended for the selection of first line EGFR TKIs. There is presently no evidence indicating superiority of TKIs when compared with conventional chemotherapy for the second or third line treatment of EGFR Wild Type NSCLC. Nonetheless, TKIs are often recommended due to their acceptable toxicities. To address this treatment dilemma, the authors performed a systematic review and meta-analysis of randomized controlled trials, comparing first-generation EGFR TKIs (TARCEVA® and IRESSA®) treatment with conventional chemotherapy, in patients with advanced NSCLC, harboring Wild Type EGFR. This pooled analysis included 1605 patients from 11 clinical trials, with EGFR Wild Type NSCLC. The primary outcome measured was Progression Free Survival (PFS) and secondary outcomes were Objective Response Rate and Overall Survival. It was noted in this analysis that conventional chemotherapy was associated with longer PFS, compared with EGFR TKIs, among patients harboring Wild Type EGFR tumors. The authors noted that there was significant PFS benefit with chemotherapy, in trials using more sensitive EGFR mutation analysis platforms, than direct Sanger sequencing, and this may be the result of identifying the “true” Wild Type EGFR tumors. The objective response rate was higher at 16.8% with chemotherapy versus 7.2% for TKIs. There was however no statistically significant difference in the overall survival between the chemotherapy and TKI groups. When subgroups of patients in these trials were analyzed, outcomes were similar regardless of line of treatment, dominant ethinicity or EGFR mutation analysis method. The lack of improvement in Overall Survival in the chemotherapy group has been attributed to the large cross over rates in the trials that were analyzed. The authors concluded that conventional chemotherapy is associated with superior Progression Free Survival and Objective Response Rates, in patients with advanced NSCLC, harboring Wild Type EGFR tumors, compared with EGFR TKIs and the present guidelines recommending EGFR TKIs in this patient group has to be reevaluated. Lee J, Hahn S, Kim D, et al. JAMA 2014;311:1430-1437
Treatment of Advanced Non–Small-Cell Lung Cancer with Epidermal Growth Factor Receptor (EGFR) Mutation or ALK Gene Rearrangement: Results of an International Expert Panel Meeting of the Italian Association of Thoracic Oncology
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society’s estimates that over 224,000 new cases of lung cancer will be diagnosed in the United States in 2014 and over 159,000 will die of the disease. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, adenocarcinoma now is the most frequent histologic subtype of lung cancer. In the mid 1990’s, following a meta-analysis of 52 randomized clinical trials, platinum based doublet chemotherapy became the accepted standard, for patients with Stage IV Non Small Cell Lung Cancer (NSCLC), after this combination demonstrated a 27% reduction in the risk of death compared to supportive care. Since then, significant advances have been made and it is now established that Platinum/Pemetrexed (ALIMTA®) combination results in superior survival in those with non squamous histology tumors whereas Platinum/ Gemcitabine (GEMZAR®) combination is superior in patients with squamous cell histology. In 2004, the discovery of Epidermal Growth Factor Receptor (EGFR) mutations in some advanced NSCLC cases with adenocarcinoma histology and the favorable responses with EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib) and IRESSA® (Gefitinib), changed the treatment paradigm in favor of targeted therapy for this patient subset. Subsequently, the discovery of rearrangements of the Anaplastic Lymphoma Kinase (ALK) gene in some patients with advanced NSCLC and adenocarcinoma histology, led to the development of agents such as XALKORI® (Crizotinib) and ZYKADIA® (Ceritinib), with promising results. It has become clear that appropriate, molecularly targeted therapy for tumors with a molecular abnormality, results in the best outcomes. This new paradigm lead to the development of evidence based recommendations by a panel of experts in thoracic oncology taking the “driver oncogenes and driver mutations” into consideration. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8% as well as other mutations in BRAF, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive and the presence of two simultaneous mutations, are rare. The following guidelines have been put together, to better manage patients with advanced adenocarcinoma of the lung. 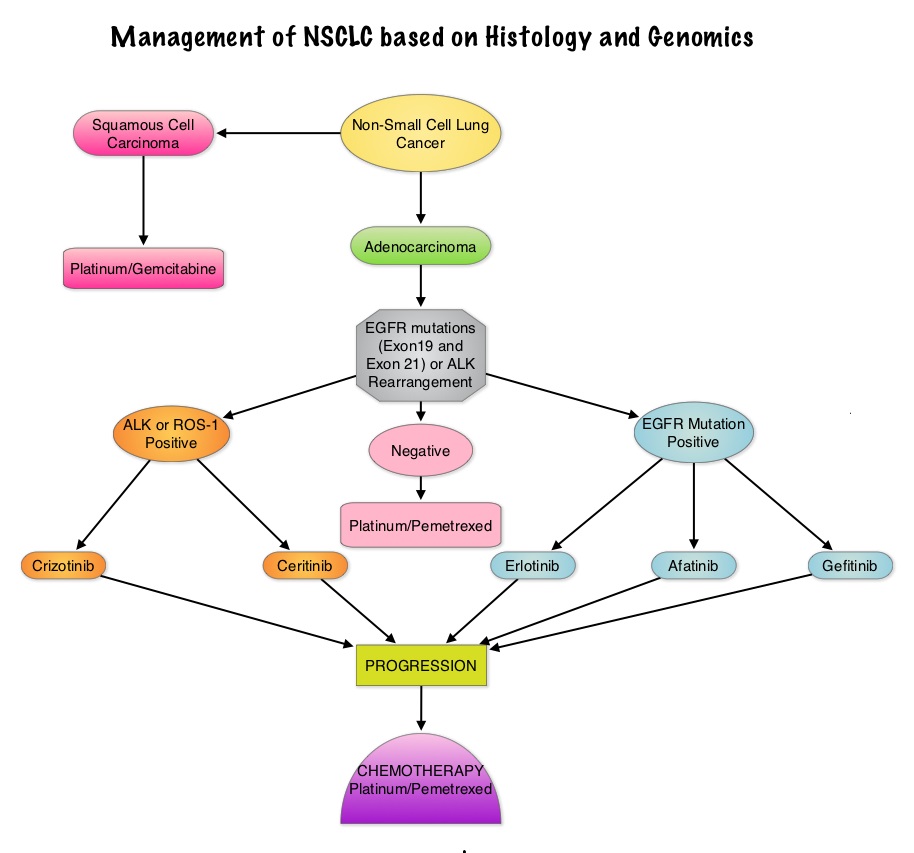
EGFR Mutations and EGFR TKIs
• All patients with advanced NSCLC with the exclusion of pure squamous cell carcinoma should be tested for EGFR mutations before first line treatment decision is made. Because EGFR mutations and ALK rearrangement are mutually exclusive, routine testing of the other is not required for those patients with one known genomic abnormality.
• Patients should be tested for the most common sensitizing mutations such as deletions in exon 19 and L858R point mutations in exon 21, as these patients clearly benefit from first line EGFR TKIs. EGFR expression by IHC (ImmunoHistoChemical) staining, EGFR gene copy by FISH (Fluorescence In Situ Hybridization) and blood based proteonomic testing by VERISTRAT® is currently not recommended for the selection of first line EGFR TKIs.
• Any one of the three available agents, TARCEVA® (Erlotinib), GILOTRIF® (Afatinib) or IRESSA® (Gefitinib) are recommended for first line treatment, as per the regulatory label, because of the absence of data on direct comparisons.
• Platinum based chemotherapy should be considered after EGFR TKI failure in eligible patients and there is no evidence to recommend a preferred chemotherapy regimen in EGFR mutation positive patients.
• If EGFR mutation positive results become available after commencement of first line chemotherapy, early interruption of chemotherapy is discouraged. However maintenance therapy with EGFR TKI should be considered after completion of first line chemotherapy.
• In patients with unknown EGFR mutational status, first line platinum based chemotherapy is the standard of care.
• Continuing EGFR TKI beyond disease progression after its use as first line treatment is not recommended.
ALK Rearrangements and Treatment Selection
• Currently, ALK status is determined by FISH technique although ALK testing by IHC analysis is gaining momentum.
• All patients with advanced NSCLC with the exclusion of pure squamous cell carcinoma should be tested for ALK rearrangement before decision about first line treatment is made. Because EGFR mutations and ALK rearrangements are mutually exclusive, routine testing of the other is not required for those patients with one known genomic abnormality.
• In the US, XALKORI® (Crizotinib) is approved for use in any line of treatment.
• Chemotherapy is allowed in any line of treatment although it is preferable to use it following treatment failure with XALKORI®.
• ZYKADIA® (Ceritinib) is approved in the US for treatment of patients with disease progression on or who are intolerant to XALKORI® (Crizotinib).
• If ALK positive results become available after commencement of first line chemotherapy, early interruption of chemotherapy is discouraged. In these patients, XALKORI® should be used as second line treatment following disease progression on chemotherapy. Maintenance therapy with XALKORI® is not recommended.
• In clinical practice, a repeat biopsy is not recommended at disease progression after treatment with EGFR TKIs or ALK inhibitors.
Gridelli C, de Marinis F, Cappuzzo F, et al. Clinical Lung Cancer 2014;15:173-181
Ceritinib in ALK-Rearranged Non–Small-Cell Lung Cancer
SUMMARY: EML4-ALK (Echinoderm Microtubule associated protein Like 4) - (Anaplastic Lymphoma Kinase) is an aberrant fusion-type oncoprotein and is a tyrosine kinase. This oncoprotein/tyrosine kinase is found in 2-7% of all Non Small Cell Lung Cancers (NSCLC) and is generated due to an inversion in the short arm of chromosome 2. This oncoprotein is more prevalent in patients with adenocarcinoma, who have little or no exposure to tobacco. Tyrosine kinases normally play an important role in cellular proliferation and differentiation. However with point mutations, translocation/rearrangement and amplification of the respective genes, the associated tyrosine kinases can potentially cause malignancy. Such is the case with mutations or translocations of the Anaplastic Lymphoma Kinase gene (ALK). XALKORI® (Crizotinib) is a small molecule Tyrosine Kinase Inhibitor that targets ALK, MET and ROS1 tyrosine kinases. In an open label phase III trial involving 347 patients with locally advanced or metastatic ALK-positive lung cancer who had received one prior platinum based regimen, treatment with XALKORI® significantly improved Progression Free Survival (PFS) and Response Rates (RR). In spite of this initial benefit, patients will however relapse within 12 months, with the average response duration of about 8 months. This has been attributed to acquired mutation within the ALK tyrosine kinase domain, amplification of the ALK fusion gene, subtherapeutic inhibition of ALK tyrosine kinase or activation of other pathways that can cause abnormal cell proliferation. Ceritinib (LDK378) is an oral, small molecule, second generation tyrosine kinase inhibitor of ALK and is 20 times as potent as XALKORI® against ALK. Unlike XALKORI®, Ceritinib does not inhibit MET kinase activity. Based on preclinical data supporting the efficacy of Ceritinib in both XALKORI® sensitive and resistant NSCLC tumors, the authors conducted a study to evaluate the antitumor activity of Ceritinib in patients with advanced NSCLC and other cancers harboring genetic alterations in ALK, in addition to determining the safety, MTD (maximum tolerated dose) and pharmacokinetics of Ceritinib. In this trial, patients who had received prior therapy with one or more ALK inhibitors as well as those with asymptomatic treated or untreated CNS metastases, were eligible to be enrolled. This study had 2 components - a dose escalation phase and an expansion phase. In the dose escalation phase, 59 patients were enrolled and the MTD of Ceritinib was determined to be 750 mg PO daily. In the expansion phase, 71 additional patients were treated for a total of 130 patients (N=59+71). Majority of these patients (94%) had advanced NSCLC. Patients with NSCLC who received at least 400mg of Ceritinib daily (N=114) had an overall response rate (RR) of 58% and median PFS was 7 months. Patients with advanced NSCLC who had received XALKORI® prior to enrollment (N=80) had a RR of 56%. The responses were noted both in patients with tumors harboring resistance mutations in the ALK tyrosine kinase domain as well as those in whom there was no new genetic alterations of ALK. Further, responses were seen in the untreated CNS lesions both in patients who had prior therapy with XALKORI® as well as those who did not. Adverse events were grade 1or 2 and GI related. These included vomiting, diarrhea, elevated aminotransferase levels and hypophosphatemia. The authors concluded that Cerifinib has marked antitumor activity in patients with advanced ALK rearranged NSCLC and in those who had progressed during XALKORI® treatment, regardless of the presence of resistance mutations in the ALK tyrosine kinase domain. Whether Cerifinib should be considered for the first line treatment of advanced ALK rearranged NSCLC, remains to be seen. Shaw AT, Kim D, Mehra R, et al. N Engl J Med 2014; 370:1189-1197
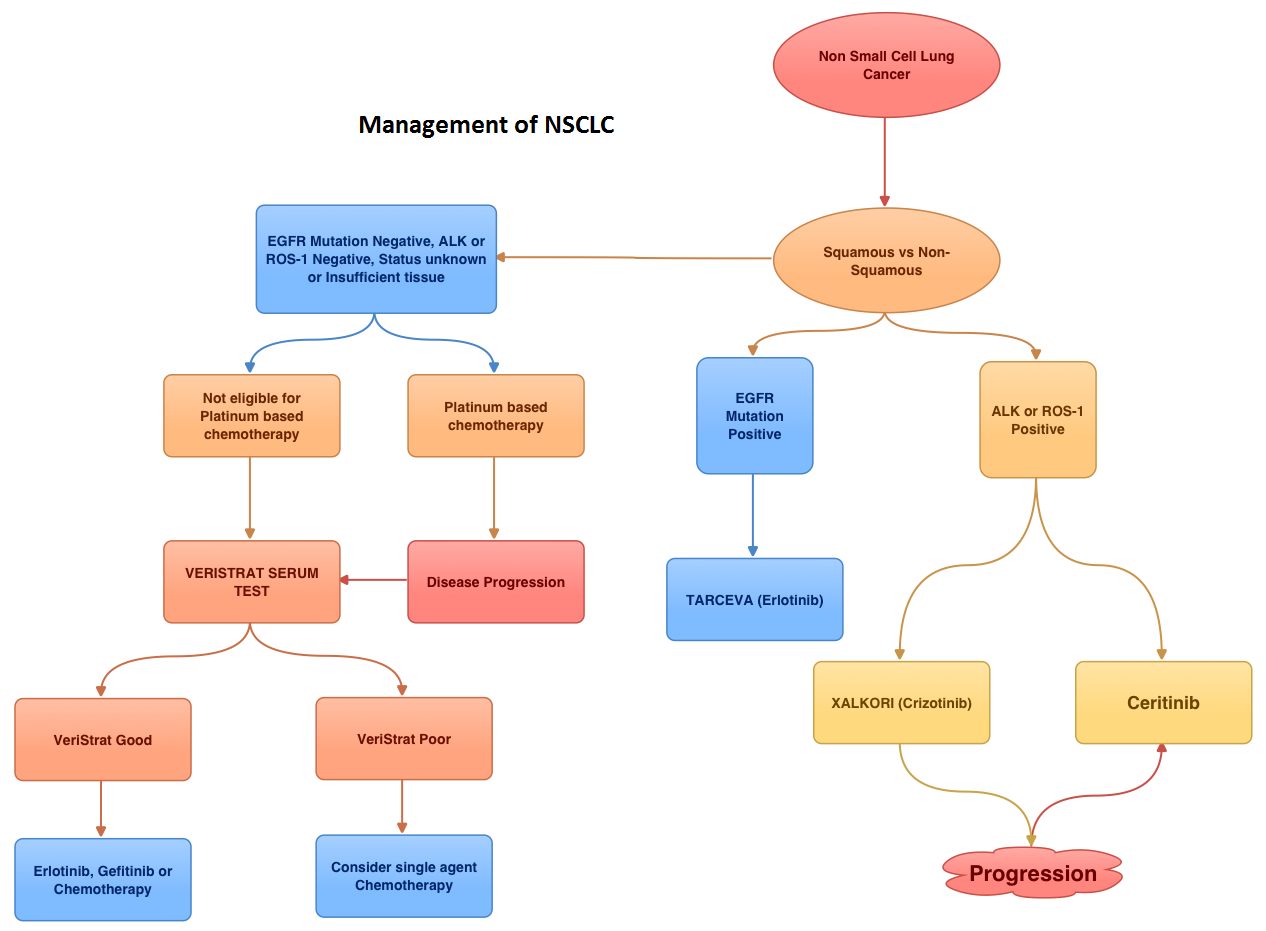
Safety and efficacy of weekly nab® paclitaxel in combination with carboplatin as first-line therapy in elderly patients with advanced non-small-cell lung cancer
SUMMARY: ABRAXANE® (Albumin-bound Paclitaxel or nab-Paclitaxel) is a solvent-free formulation of Paclitaxel with a superior therapeutic index and delivers higher concentrations of the drug’s active ingredient into the tumor cell. This is unlike solvent based taxanes such as TAXOL® (Paclitaxel) and TAXOTERE® (Docetaxel), which have delivery vehicles such as Cremaphor and Polysorbate 80 respectively. By virtue of being solvent free, ABRAXANE® can be administered over a shorter period of time without premedications and is associated with fewer side effects with possibly superior efficacy. In a phase III trial, 1052 treatment naive patients with Stage IIIB/IV Non Small Cell Lung Cancer (NSCLC) were randomly assigned to receive ABRAXANE® 100 mg/m2 weekly and PARAPLATIN® (Carboplatin) at Area Under the Concentration-time curve (AUC) 6, once every 3 weeks (nab-PC) or TAXOL® 200mg/m2 plus PARAPLATIN® AUC 6 once every 3 weeks (sb-PC). Patients were stratified by disease (Stage IIIB vs IV), age (< 70 vs ≥ 70 years), sex (male vs female), histology (squamous vs adenocarcinoma vs others). The primary end point was Overall Response Rate (ORR). Secondary end points included Progression Free Survival (PFS) and Overall Survival (OS). In their original report, the authors concluded that the study met its primary end point and ABRAXANE® combination (nab-PC) significantly improved ORR compared to TAXOL® combination (sb-PC) and was also associated with less neuropathy. In a sub-set analysis of patients 70 years or older (N=156), those in the ABRAXANE® group had a significantly longer median OS compared to the TAXOL® group (19.9 vs 10.4 months, HR=0.58, P=0.009). The PFS in this elderly group trended in favor of ABRAXANE® (8 vs 6.8 months, P=0.13). This survival benefit was not seen in the younger patients. Elderly patients with NSCLC usually tend to have other co-morbidities and treatment can be challenging. With lower incidence of toxicities such as neuropathy, neutropenia and arthralgias, ABRAXANE® combination therapy can be a valuable option for the first line treatment of elderly patients with advanced NSCLC of all histologies. Socinski MA, Langer CJ, Okamoto I, et al. Ann Oncol. 2013;24:314-321
Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer
SUMMARY: EML4-ALK (Echinoderm Microtubule associated protein Like 4) - ALK (Anaplastic Lymphoma Kinase) is an aberrant fusion-type oncoprotein and is a tyrosine kinase. This oncoprotein/tyrosine kinase is found in 2-7% of all Non Small Cell Lung Cancers (NSCLC) and is generated due to an inversion in the short arm of chromosome 2. This oncoprotein is more prevalent in patients with adenocarcinoma, who have little or no exposure to tobacco. Tyrosine kinases normally play an important role in cellular proliferation and differentiation. However with point mutations, translocation/rearrangement and amplification of the respective genes, the associated tyrosine kinases can potentially cause malignancy. Such is the case with mutations or translocations of the Anaplastic Lymphoma Kinase gene (ALK). Crizotinib (XALKORI®) is a small molecule Tyrosine Kinase Inhibitor that targets ALK, MET and ROS1 tyrosine kinases. In this open label phase III trial, 347 patients with locally advanced or metastatic ALK-positive lung cancer who had received one prior platinum based regimen, were randomly assigned to receive XALKORI® 250 mg PO twice daily (N=173) or intravenous chemotherapy with either Pemetrexed (ALIMTA®) 500 mg/m2 or Docetaxel (TAXOTERE®) 75 mg/m2, every 3 weeks (N=174). The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Overall Survival (OS), Response Rate (RR) and safety. The median PFS was 7.7 months in the XALKORI® group as compared to 3 months in the chemotherapy group (HR=0.49; P<0.001). The Response Rates for XALKORI® and chemotherapy were 65% and 20% respectively (P<0.001). At the time of interim analysis, there was no significant difference in the OS between the XALKORI® and chemotherapy groups. The authors pointed out that this lack of OS benefit was due to the high cross over rate to the XALKORI® group from the chemotherapy group. Patients in the XALKORI® group had better quality of life, greater reduction in lung cancer symptoms and were on the study treatment longer, than with chemotherapy. The authors concluded that XALKORI® improves PFS, Response Rates as well as Quality Of Life in patients with previously treated, ALK positive advanced Non Small Cell Lung Cancer. This is remarkable, considering that the response rates in this patient population treated with second line chemotherapy is around 10-15%. As we move forward, it is very likely that tailored therapy based on molecular genotyping will become standard practice. Shaw AT, Kim D, Nakagawa K, et al. N Engl J Med 2013; 368:2385-2394
Results of Initial Low-Dose Computed Tomographic Screening for Lung Cancer
SUMMARY: The National Lung Screening Trial (NLST), a federally funded U.S. study, enrolled 53,439 asymptomatic participants, 55 to 74 years of age, with at least 30 pack-year smoking history, and were randomized to undergo annual screening with either low dose CT scan (n=26,715) or a chest X-Ray (n=26,724), for three years. The use of low dose CT scans for 3 years in this high risk, healthy patients, resulted in a 20% reduction in Lung Cancer mortality, compared to screening with a chest X-Ray. Based on these findings, Lung Cancer Screening is recommended for the following groups
1) People 55-74 years of age with no signs or symptoms of Lung Cancer
2) Current or former smoker with a 30 pack year smoking history (Number of years smoked multiplied by the number of packs of cigarettes per day)
3) Current smokers are strongly urged to enter a smoking cessation program
4) Former smokers must have quit smoking within the past 15 years
Lung Cancer screening is performed using a non-contrast, low dose CT scan. People with serious co-morbid conditions, those on home oxygen and individuals with metallic devices or implants in the chest or back (which can interfere with the scan) should be excluded from Lung Cancer screening. It should be noted that Lung cancer screening with low dose CT scan is presently not covered by most insurance plans. The National Lung Screening Trial Research Team. N Engl J Med 2013;368:1980-1991
PROSE: Randomized proteomic stratified phase III study of second line erlotinib versus chemotherapy in patients with inoperable non–small cell lung cancer (NSCLC)
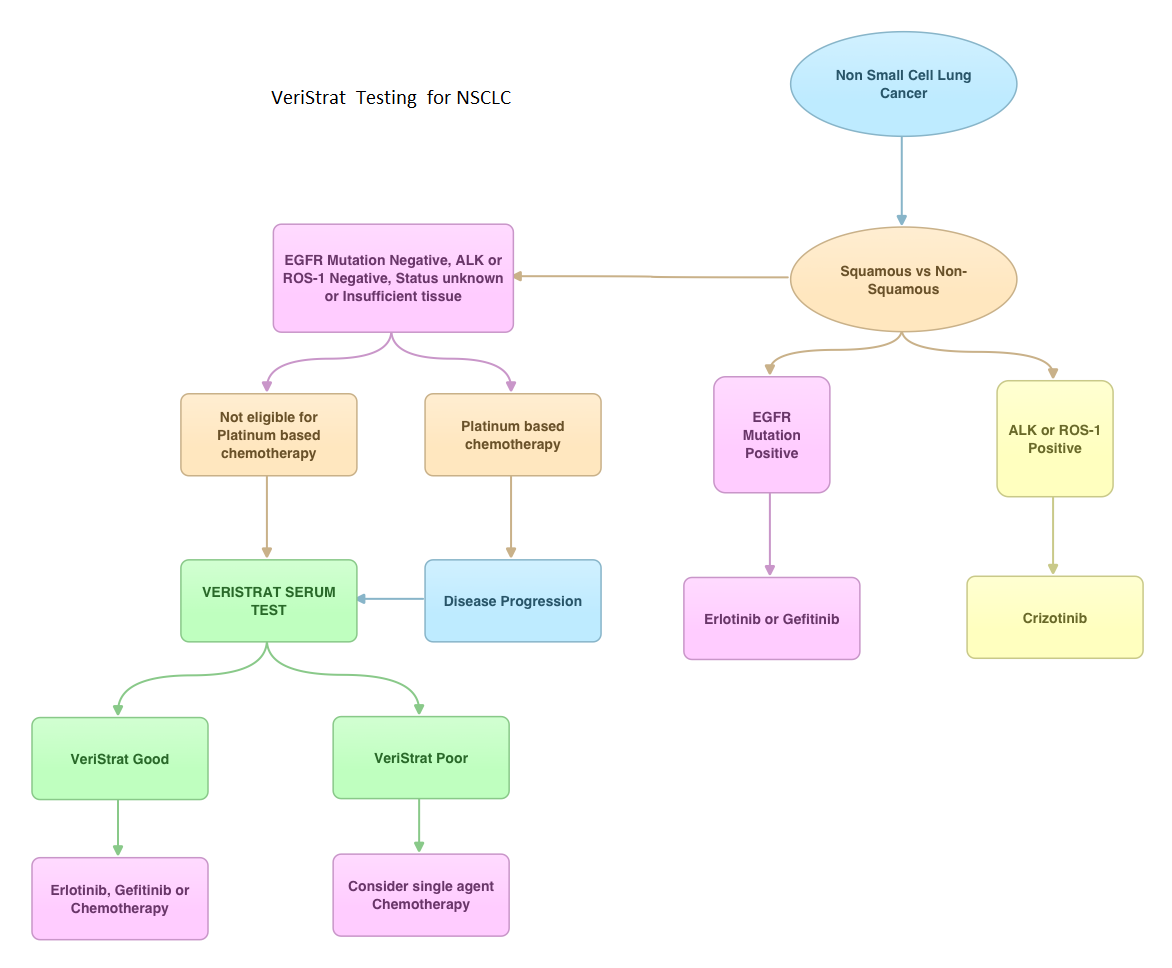
SUMMARY: VeriStrat ® is a clinically validated serum/plasma-based assay, for patients with advanced Non Small Cell Lung Cancer (NSCLC). VeriStrat® is a serum test of prognostic and predictive value that classifies patients as VeriStrat-Good (VS-G) or VeriStrat-Poor (VS-P) based on eight mass spectral peaks or proteomic patterns of the patients serum. Proteomics is the large-scale study of protein structure and functions. VeriStrat® testing is protein based and therefore has no correlation with known genomic biomarkers. It is well established that EGFR-TKIs (Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors) are more effective in NSCLC patients with EGFR activating mutations. PROSE is a multicenter, double blind, randomized, VeriStrat® stratified, phase III study. In this trial, over 90% of the patients had no EGFR mutations (EGFR-Wild Type). Two hundred and eighty five (285) patients with advanced NSCLC who had first line treatment regimen with platinum-based therapy were randomly assigned to receive second line chemotherapy (CT) with single agent ALIMTA® (Pemetrexed) or TAXOTERE® (Docetaxel), at standard doses (N=129) or TARCEVA® (Erlotinib) 150 mg po qd (N=134). Patients and study investigators were blinded to the patients VeriStrat® status. Patients were classified as VeriStrat-Good or VeriStrat-Poor based on the VeriStrat® results. Patients in the treatment groups were stratified by age, gender, tumor histology, ECOG-PS and smoking history. Crossover was permitted upon disease progression. The primary objective of the study was to demonstrate differential treatment benefit between TARCEVA® and CT with regards to Overall Survival (OS). Median overall survival (OS) was 9 months for the patients in the CT group and 7.7 months for TARCEVA® group and this was not statistically significant (P=0.3). However when evaluated by VeriStrat® status, CT was beneficial for the VeriStrat-Poor patients compared to TARCEVA®, with significantly better median OS (6.3 vs 3 months, P=0.02). Age, gender, histology (squamous vs non-squamous) and smoking history had no impact on the overall survival. The authors concluded that patients classified as VeriStrat-Poor have better survival with CT than TARCEVA®, whereas patients classified as VeriStrat-Good have similar survival with TARCEVA® and CT. VeriStrat® testing therefore, can help physicians choose between TARCEVA® and CT, for their patients with advanced NSCLC. This test helps physicians identify patients who are likely to have good or poor outcomes after treatment with EGFR inhibitors and thereby can provide valuable insight into whether CT or targeted therapy with TARCEVA®, a EGFR-TKI, is appropriate for their patients with advanced NSCLC, in the second line setting. This information is especially important for patients without an EGFR mutation or for those, whose EGFR mutation status is unknown. Sorlini C, Barni S, Petrelli F, et al. J Clin Oncol 29: 2011 (suppl; abstr TPS214)
Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations
SUMMARY: GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. The approval of GILOTRIF® was based on a multi-center, international, open-label, randomized, phase III trial, in which 345 patients with Stage IIIB (wet)/IV lung adenocarcinoma, with tumors demonstrating Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations, as detected by an FDA-approved test, were enrolled in a 2:1 ratio. Patients were randomized to receive GILOTRIF® 40 mg orally once daily (n=230) or ALIMTA® (Pemetrexed)/Cisplatin (n=115) given every 21 days for up to six cycles. Patients were stratified according to EGFR mutation status (exon 19 deletion vs. exon 21 L858R vs. ‘other’) and race (Asian vs. non-Asian). The primary endpoint was Progression Free Survival (PFS). The median PFS in the GILOTRIF® group was 11.1 months and 6.9 months in the chemotherapy group (HR= 0.58, P<0.001). In patients whose tumors demonstrated EGFR mutations, the median PFS was 13.6 months in the GILOTRIF® arm and 6.9 months in the chemotherapy arm (HR= 0.47, P<0.001). Objective response rates were 56% and 23% in the GILOTRIF® and chemotherapy groups respectively (P=0.001). There was no statistically significant difference in overall survival between the two treatment groups. The most frequent adverse reactions in the GILOTRIF® group were skin rash, pruritus, stomatitis, diarrhea and decreased appetite. The authors concluded that GILOTRIF® is better than chemotherapy in the first line treatment of EGFR mutant Non Small Cell Lung Cancer patients. However, it remains to be seen if this agent is superior to TARCEVA® (Erlotinib) and IRESSA® (Gefitinib). Sequist LV, Yang JC, Yamamoto N, et al. J Clin Oncol 2013;31:3327-3334
LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations.
SUMMARY: GILOTRIF® (Afatinib) is an oral, irreversible blocker of the ErbB family which includes EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. The approval of GILOTRIF® was based on a multi-center, international, open-label, randomized, phase III trial, in which 345 patients with Stage IIIB (wet)/IV lung adenocarcinoma, with tumors demonstrating Epidermal Growth Factor Receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations, as detected by an FDA-approved test, were enrolled in a 2:1 ratio. Patients were randomized to receive GILOTRIF® 40 mg orally once daily (n=230) or ALIMTA® (Pemetrexed)/Cisplatin (n=115). Patients were stratified according to EGFR mutation status (exon 19 deletion vs. exon 21 L858R vs. ‘other’) and race (Asian vs. non-Asian). The primary endpoint was Progression Free Survival (PFS). The median PFS in the GILOTRIF® group was 11.1 months and 6.9 months in the chemotherapy group (HR= 0.58, P<0.001). In patients whose tumors demonstrated EGFR mutations, the median PFS was 13.6 months in the GILOTRIF® arm and 6.9 months in the chemotherapy arm (HR= 0.47, P<0.0001). Objective response rates were 50.4% and 19.1% in the GILOTRIF® and chemotherapy groups respectively. There was no statistically significant difference in overall survival between the two treatment groups. The most frequent adverse reactions in the GILOTRIF® group were skin rash, pruritus, stomatitis, diarrhea and decreased appetite. The authors concluded that GILOTRIF® is better than chemotherapy in the first line treatment of EGFR mutant Non Small Cell Lung Cancer patients. However, it remains to be seen if this agent is superior to TARCEVA® (Erlotinib) and IRESSA® (Gefitinib). Yang JC, Shuler M, Yamamoto N, et al. J Clin Oncol 2012;30(18,Suppl):abstract LBA 7500.
Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening
Summary: The National Lung Screening Trial (NLST) enrolled 53,454 individuals at high risk for lung cancer and randomly assigned them to undergo three annual screenings with either non-contrast, low-dose CT scan (LDCT) or single-view posteroanterior chest X-ray. This study demonstrated a 20% reduction in the risk of death from lung cancer in the group of people who were screened with LDCT compared to those who were screened with chest X-ray (P=0.004). Based on this study and data, the American Cancer Society has recommended lung cancer screening for individuals 55 to 74 years of age who are in fairly good health, have at least a 30 pack-year smoking history and are either still smoking or have quit smoking within the past 15 years. If a person remains in good health, annual LDCT is continued until the age of 74. Smokers should be counseled to quit smoking. Further more, people with abnormal scans should receive appropriate care and follow up. Lung cancer screening with LDCT is presently not covered by most insurance plans. The National Lung Screening Trial Research Team. N Engl J Med 2011; 365:395-409
LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations.
SUMMARY: Afatinib is an oral tyrosine kinase inhibitor that irreversibly inhibits EGFR (ErbB1), HER2 (ErbB2), and ErbB4. In this study, chemo naïve patients with advanced stage adenocarcinoma of the lung (stage IIIB/IV) with EGFR mutations, were randomly assigned to receive either Afatinib or a combination of Pemetrexed and Cisplatin. Primary endpoint was progression-free survival (PFS). Treatment with Afatinib resulted in a significantly prolonged PFS compared to combination chemotherapy (median 11.1 vs 6.9 months; HR 0.58; P=0.0004). Further, patients in the Afatinib group had a higher objective response rate ((56% vs 23%; p<0.0001), as well as significant delay in symptom progression, compared to the combination chemotherapy group. Yang JC-H, Schuler MH, Yamamoto N, et al. J Clin Oncol. 2012;30(suppl; abstr LBA7500).
Anaplastic Lymphoma Kinase Inhibition in Non Small Cell Lung Cancer
SUMMARY: The therapeutic target of interest is an aberrant fusion gene, EML4-ALK. EML4 (echinoderm microtubule-associated protein-like 4) - ALK (anaplastic lymphoma kinase) is a fusion-type oncoprotein and is tyrosine kinase. This oncoprotein/tyrosine kinase is found in 2-7% of all Non Small Cell Lung Cancers (NSCLC) and is generated due to an inversion in the short arm of chromosome 2. This oncoprotein is more prevalent in patients with adenocarcinoma who have little or no exposure to tobacco. Tyrosine kinases normally play an important role in cellular proliferation and differentiation. However with point mutations, translocation/rearrangement and amplifications of their respective genes, these tyrosine kinases can potentially cause malignancy. Such is the case with mutations or translocations of the Anaplastic Lymphoma Kinase gene (ALK). In an article published in the October 28, 2010 issue of the NEJM, Crizotinib an oral small molecule tyrosine kinase inhibitor of ALK tyrosine kinase resulted in an overall response rate of 57% in patients who had progressed on prior therapies. Stable disease was noted in 33% of the patients. This is remarkable considering that the response rates in this patient population treated with second line chemotherapy is around 10-15%. As we move forward, it is very likely that genotyping patients and tailoring therapy accordingly, will become standard practice. N Engl J Med 2010; 363:1693-1703
Results of a randomized, phase III trial of nab-paclitaxel (nab-P) and carboplatin (C) compared with cremophor-based paclitaxel (P) and carboplatin as first-line therapy in advanced non-small cell lung cancer (NSCLC).
SUMMARY: In this randomized phase III trial, the efficacy of nab-paclitaxel (ABRAXANE®) and carboplatin was compared with paclitaxel and carboplatin in advanced NSCLC of all histologic types. Patients enrolled were chemonaïve, with stage IIIb and stage IV non small cell lung cancer. Five hundred and twenty one (521) received ABRAXANE® without any premedications at 100 mg/m2 on days 1, 8 and 15 along with carboplatin given on day 1 at an AUC of 6. The control group of 525 patients received standard Paclitaxel 200mg/m2 and carboplatin at an AUC of 6 on day 1. The primary end point was overall response rate. The ABRAXANE® group had a response rate of 33% compared to 25% for the standard paclitaxel group. When broken down by histology, the response rates in those with squamous cell carcinoma was 41% in the ABRAXANE® group versus 24% in the standard paclitaxel arm and the non squamous subtypes had a response rate of 26% versus 25% in the ABRAXANE® and paclitaxel group respectively. It is hypothesized that the superior response rates in squamous cell histology may be due to the overexpression by this sub type of an albumin receptor called Caveolin–1. ABRAXANE® which is an albumin bound paclitaxel utilizes the albumin receptor Caveolin-1 (CAV1) pathway and thereby may achieve a higher intratumoral drug concentration. J Clin Oncol 28:18s, 2010 (suppl; abstr LBA7511)