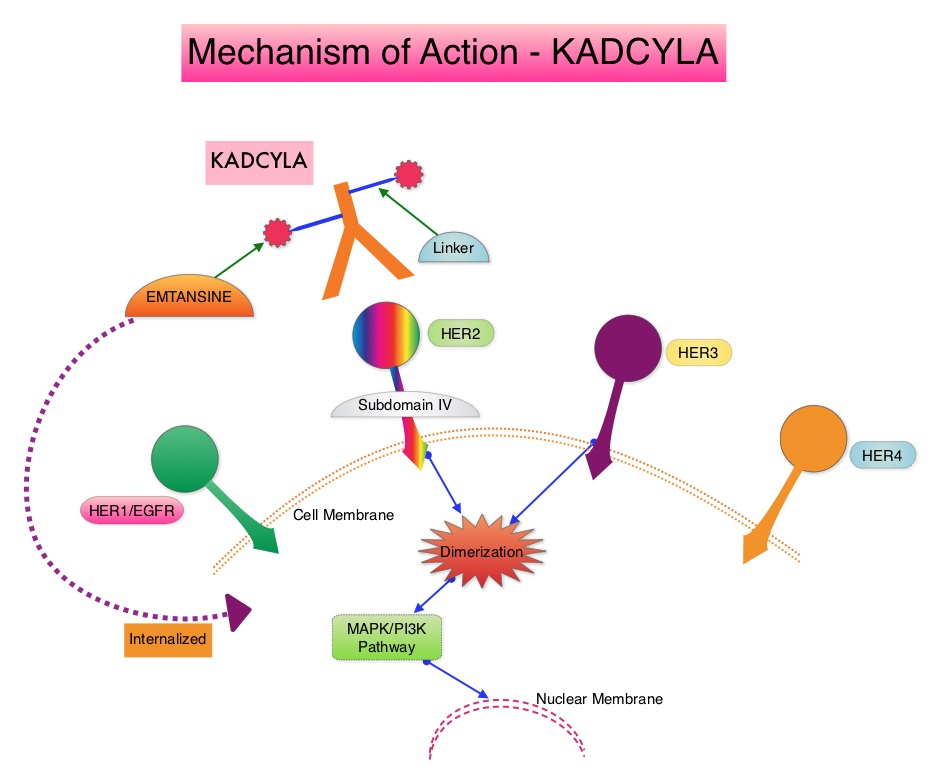
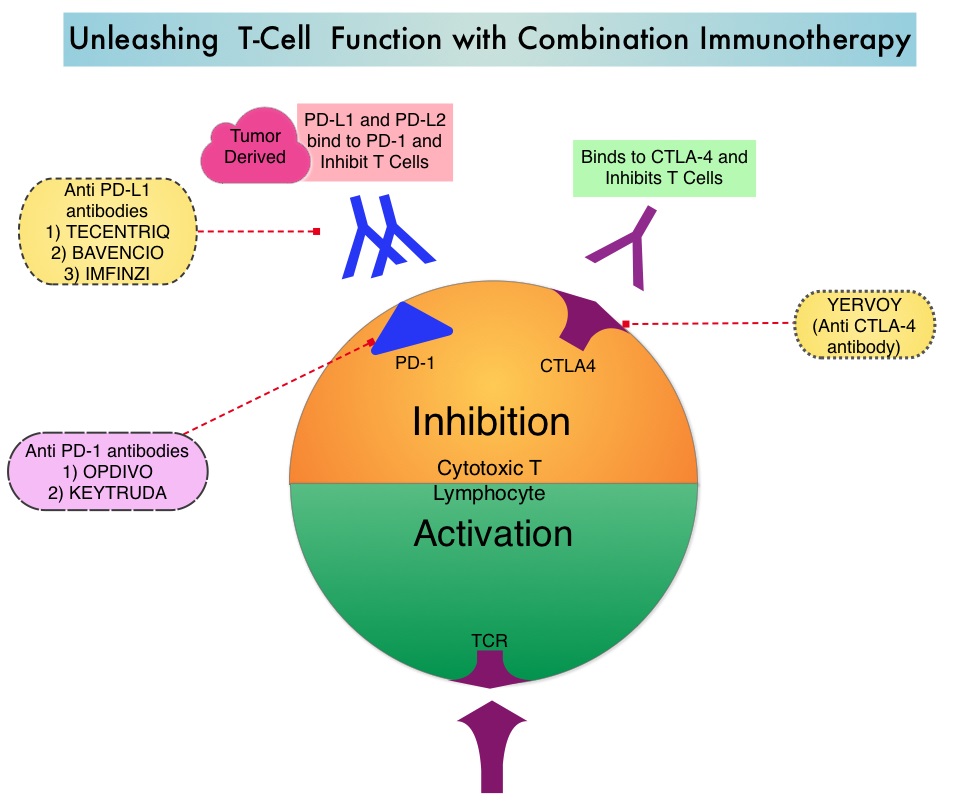
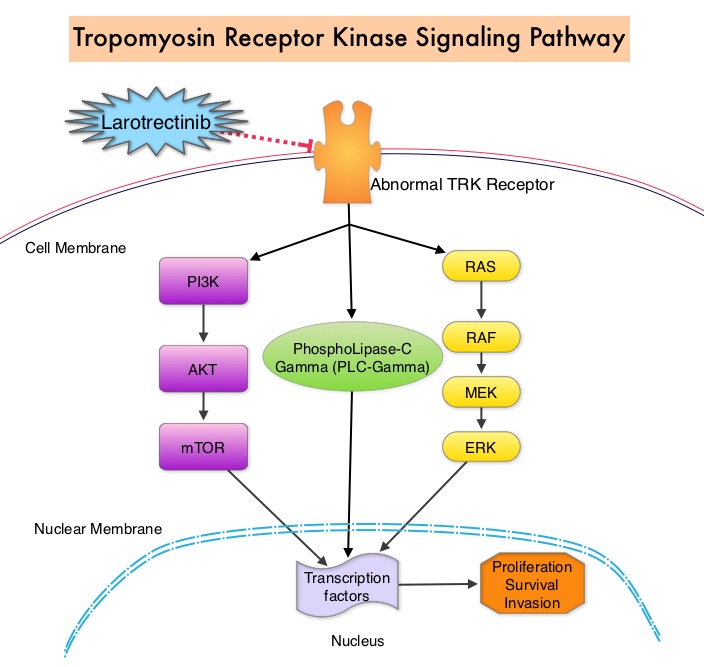
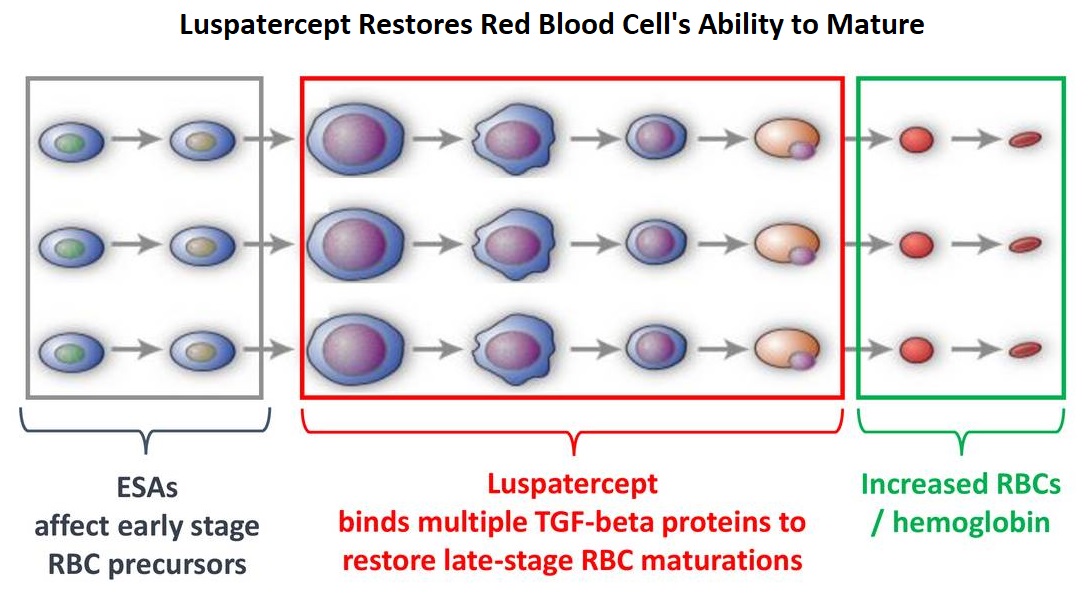
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 30,770 new cases will be diagnosed in 2018 and 12,770 patients are expected to die of the disease. Multiple Myeloma (MM) in 2018 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). REVLIMID® (Lenalidomide) based regimens are often prescribed for patients with newly diagnosed, transplant-ineligible Multiple Myeloma. REVLIMID®, a thalidomide analogue has immunomodulatory, tumoricidal, and antiangiogenic properties, and synergizes with Dexamethasone to enhance anti-myeloma activity. DARZALEX® (Daratumumab) is a human IgG1 monoclonal antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Mediated Cytotoxicity and direct apoptosis. Additionally, DARZALEX® may have a role in immunomodulation by depleting CD38-positive regulator Immune suppressor cells, and thereby expanding T cells, in patients responding to therapy.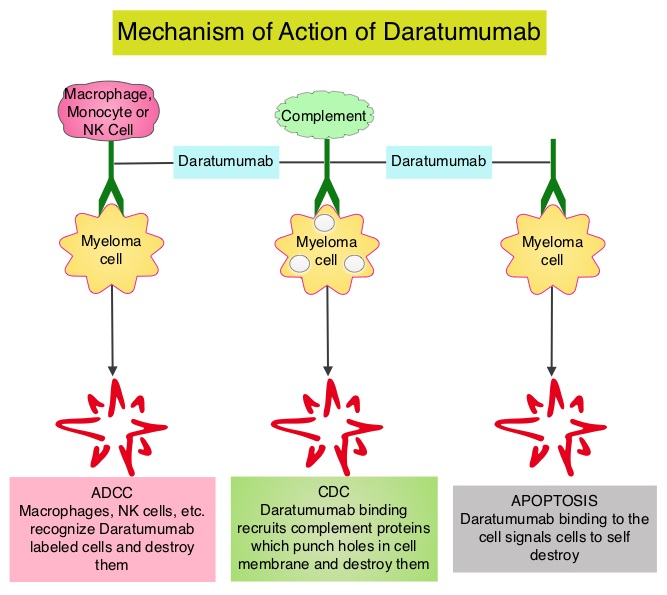
The FDA in May, 2018 approved DARZALEX® in combination with VELCADE® (Bortezomib), a proteasome inhibitor, Melphalan, an alkylating agent and Prednisone (VMP regimen), for the treatment of patients with newly diagnosed Multiple Myeloma who are ineligible for Autologous Stem Cell Transplant (ASCT). VMP regimen however is mostly utilized in Europe and not in the US. In the POLLUX trial, addition of DARZALEX® to REVLIMID® and Dexamathasone (D-Rd) showed the greatest benefit, with a 63% reduction in risk of disease progression or death (HR=0.37; P<0.001) in patients with Multiple Myeloma who had at least one prior line of therapy, compared to REVLIMID® and Dexamathasone (Rd) . Based on the efficacy and tolerable safety profile of D-Rd, the authors conducted a phase III study (MAIA), comparing D-Rd to Rd in transplant-ineligible newly diagnosed Multiple Myeloma patients and reported the prespecified interim analysis of the study.
The MAIA study is a multicenter, international, open-label, phase III trial, which included 737 newly diagnosed Myeloma patients who were not candidates for high-dose chemotherapy and Autologous Stem Cell Transplant (ASCT), due to age 65 years or older or comorbidities. Patients were randomly assigned 1:1 to receive REVLIMID® 25 mg orally on days 1-21 of each 28-day cycle and Dexamethasone 40 mg once a week, with or without DARZALEX®. Patients assigned DARZALEX® (D-Rd regimen) received 16 mg/kg weekly for the first 8 weeks (cycles 1 and 2), every other week for 16 weeks (cycles 3 to 6), and then every 4 weeks (cycle 7 and beyond) until disease progression or unacceptable toxicity. Treatment groups were well balanced. The median patient age was 73 years and only 1% of patients were 65 years of age or less whereas 44% of patients were 75 years or older. Cytogenetic risk level could be determined in 642 patients of the total population. Eighty-six percent (86%) of these patients were standard risk and 14% were considered high risk. The Primary end point was Progression Free Survival (PFS). Key Secondary endpoints included Overall Response Rate (ORR), Minimal Residual Disease (MRD) negativity rate (10-5 sensitivity), and Safety.
The prespecified interim analysis occurred with a median follow up of 28 months. DARZALEX® regimen significantly improved PFS with the median PFS not reached with D-Rd compared with 31.9 months in the Rd group (HR=0.55; P<0.0001). This represented a 45% reduction in the risk of progression or death in patients treated with D-Rd. D-Rd also resulted in deeper responses with a Complete Response (CR) or better rate of 47.6% in the D-Rd group compared with 24.7% in the Rd group (P<0.0001). The Very Good Partial Response (VGPR) or better rate was 79.3% in the D-Rd arm compared with 53.1% in the Rd arm (P<0.0001). The MRD-negative rate was more than threefold higher with D-Rd versus Rd at 24% versus 7%, respectively. Higher rates of neutropenia, and leukopenia were observed in the D-Rd arm and the safety profile was consistent with previously reported DARZALEX® studies.
The authors concluded that the addition of DARZALEX®, to REVLIMID® and Dexamethasone significantly reduced the risk of progression or death by 45% in newly diagnosed Multiple Myeloma patients who are transplant-ineligible and these results support D-Rd as a new standard of care for this patient group. Phase 3 Randomized Study of Daratumumab Plus Lenalidomide and Dexamethasone (D-Rd) Versus Lenalidomide and Dexamethasone (Rd) in Patients with Newly Diagnosed Multiple Myeloma (NDMM) Ineligible for Transplant (MAIA). Facon T, Kumar SK, Plesner T, et al. Presented at: ASH Annual Meeting and Exposition; December 4-8, 2018; San Diego, California. Abstract LBA-2.