
The FDA on March 22, 2018 approved TASIGNA® for pediatric patients 1 year of age or older with newly diagnosed Philadelphia chromosome positive Chronic Myeloid Leukemia in Chronic Phase (Ph+ CML-CP) or Ph+ CML-CP resistant or intolerant to prior Tyrosine Kinase Inhibitor (TKI) therapy. TASIGNA® is a product of Novartis Pharmaceuticals Corporation.
Month: April 2018
ADCETRIS® (Brentuximab vedotin)
The FDA on March 20, 2018 approved ADCETRIS® to treat adult patients with previously untreated stage III or IV classical Hodgkin lymphoma (cHL), in combination with chemotherapy. ADCETRIS® is a product of Seattle Genetics, Inc.
Frontline TECENTRIQ® along with AVASTIN® and Chemotherapy Improves Survival in Advanced NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. Immunotherapy is an accepted second line intervention after platinum-based chemotherapy in patients with advanced NSCLC, and is an approved first line therapy, for patients with high PD-L1 expressing tumors (50% or more). Further, immunotherapy with KEYTRUDA® (Pembrolizumab), in combination with chemotherapy, has been approved for first line treatment of patients with advanced non-squamous NSCLC, irrespective of PD-L1 expression.
TECENTRIQ® (Atezolizumab) is an anti-PDL1 monoclonal antibody, designed to directly bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, thereby blocking its interactions with PD-1 and B7.1 receptors and thus enabling the activation of T cells. TECENTRIQ® was approved by the FDA in October 2016 for the treatment of patients with metastatic Non Small Cell Lung Cancer (NSCLC) whose disease progressed during or following Platinum-containing chemotherapy. AVASTIN® (Bevacizumab) is a biologic antiangiogenic antibody, directed against Vascular Endothelial Growth Factor (VEGF), and prevents the interaction of VEGF to its receptors (Flt-1 and KDR) on the surface of endothelial cells. The interaction of VEGF with its receptors has been shown to result in endothelial cell proliferation and new blood vessel formation. Combining TECENTRIQ® and AVASTIN® is supported by the following scientific rationale. AVASTIN® in addition to its established anti-angiogenic effects, may further enhance the ability of TECENTRIQ® to restore anti-cancer immunity, by inhibiting VEGF-related immunosuppression, promoting T-cell tumor infiltration and enabling priming and activation of T-cell responses against tumor antigens.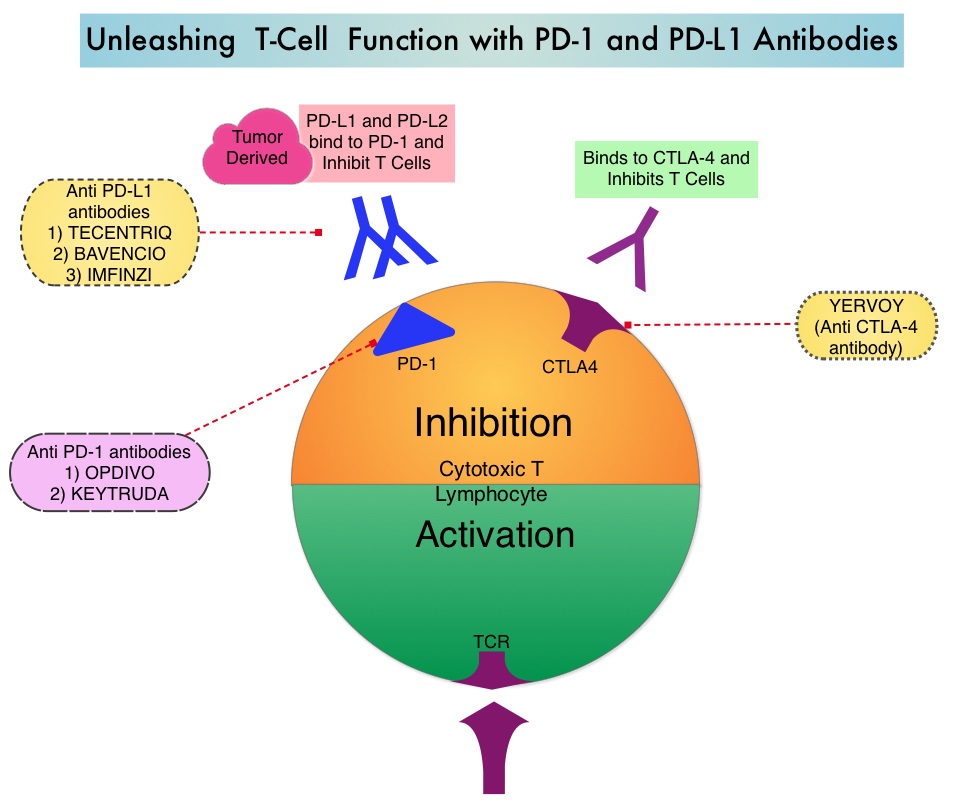
IMpower150 is a multicenter, open-label, randomized, phase III study, conducted to evaluate the efficacy and safety of TECENTRIQ® in combination with Carboplatin and Paclitaxel with or without AVASTIN®, in patients with stage IV, treatment naïve, non-squamous NSCLC. This study enrolled 1,202 patients, who were randomized (1:1:1) to receive either TECENTRIQ® along with Carboplatin and Paclitaxel (Group A), TECENTRIQ® and AVASTIN® along with Carboplatin and Paclitaxel (Group B), or AVASTIN® plus Carboplatin and Paclitaxel (Group C – control arm). During the treatment-induction phase, patients in Group A received TECENTRIQ® 1200 mg IV along with Carboplatin AUC 6 and Paclitaxel 200mg/m2 IV on Day 1 of a 3-week treatment cycle for 4 or 6 cycles. Following the induction phase, patients received maintenance treatment with TECENTRIQ® on the same dose schedule until disease progression. Patients in Group B received AVASTIN® 15 mg/kg IV, along with TECENTRIQ®, Carboplatin and Paclitaxel IV, Day 1 of a 3-week treatment cycle for 4 or 6 cycles followed by maintenance treatment with the TECENTRIQ® and AVASTIN® until disease progression. Patients in the control Group C received AVASTIN® plus Carboplatin and Paclitaxel every 3 weeks for 4 or 6 cycles followed by AVASTIN® maintenance treatment until disease progression. Patients with tumors demonstrating ALK and EGFR mutations were excluded from the primary Intention-To-Treat (ITT) analysis. Patients were also tested for a tumor T-effector gene expression signature (based on phase II trial finding of prolonged Overall Survival in patients with high gene expression signature levels, treated with TECENTRIQ®). The median age was 63 years and the minimum follow up at the time of the analysis was 9.5 months. For the interim analysis, the study was only designed to compare Groups B and C. The co-Primary endpoints were Progression Free Survival (PFS) and Overall Survival in the Intention-to-Treat (ITT) population comparing patients in Group B and C. These end points were also evaluated in subgroup of people who had a specific biomarker (T-effector gene signature expression).
It was noted that at this interim analysis, the combination of TECENTRIQ® and AVASTIN® plus Carboplatin and Paclitaxel, significantly improved PFS and reduced the risk of disease worsening or death by 38% (HR=0.62; P<0.0001), compared to AVASTIN® plus Carboplatin and Paclitaxel alone. This PFS benefit was observed across key subgroups, regardless of PD-L1 expression status, including PD-L1–negative patients (HR 0.77). Further, the median PFS in the population of patients with defined expression of a T-effector gene signature expression in the tumor tissue, was 11.3 months versus 6.8 months (HR 0.51; P<0.0001). Roche on March 26, 2018 announced that the IMpower150 study met its co-primary endpoint of Overall Survival as well. Details will soon become available.
It was concluded that combining chemotherapy with immunotherapy and antiangiogenic agents significantly improved PFS as well as Overall Survival, in patients with treatment naïve, advanced non-squamous NSCLC. This strategy can completely eliminate the need for patient selection based on a particular biomarker, and could benefit larger number of patients with advanced NSCLC. Reck M. Primary PFS and safety analyses of a randomized Phase III study of carboplatin + paclitaxel +/− bevacizumab, with or without atezolizumab in 1L non-squamous metastatic NSCLC (IMpower150). Annals of Oncology, 2017;28(11). Abstract LBA1_PR. https://www.roche.com/media/store/releases/med-cor-2018-03-26.htm
FDA Approves RUBRACA® for Maintenance Treatment of Recurrent Ovarian Cancer
SUMMARY: The FDA on April 6, 2018, approved RUBRACA® (Rucaparib), a Poly ADP-Ribose Polymerase (PARP) inhibitor, for the maintenance treatment of recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a Complete or Partial Response to platinum-based chemotherapy. RUBRACA® was initially approved in December 2016 as monotherapy for the treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer, who have been treated with two or more chemotherapies.
RUBRACA® is an oral, small molecule inhibitor of Poly-Adenosine diphosphate [ADP] Ribose Polymerase (PARP), developed for treatment of ovarian cancer, associated with Homologous Recombination DNA repair deficiency (HRD). Previously published clinical data had suggested that ovarian cancer patients with and without evidence of a germline BRCA mutation, benefit from treatment with a PARP inhibitor, and that maintenance treatment with a PARP inhibitor following a response to platinum-based treatment increases Progression Free Survival (PFS), in patients with ovarian cancer. Even though patients with or without BRCA mutation benefited, the most benefit was derived in those with BRCA mutation.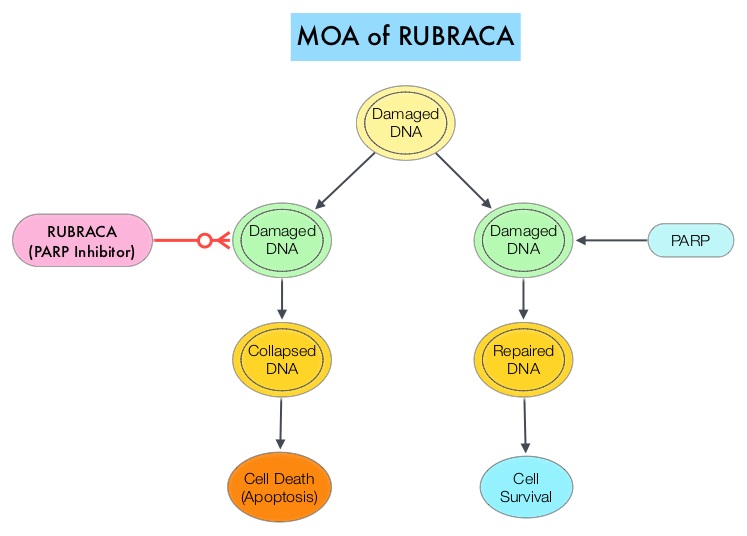
The approval of RUBRACA® was based on ARIEL3, a randomized, double-blind, placebo-controlled, phase III trial, which evaluated the benefit of RUBRACA® versus placebo, after response to second-line or later platinum-based chemotherapy, in patients with high-grade, recurrent, platinum-sensitive ovarian carcinoma. In this trial, 561 patients were randomly assigned in a 2:1 ratio to receive RUBRACA® 600 mg orally twice daily (N=372) or placebo (N=189). Treatment was continued until disease progression or unacceptable toxicity. Eligible patients had recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, and had been treated with at least two prior treatments of platinum-based chemotherapy, and were in Complete or Partial Response to the most recent platinum-based chemotherapy. Patients had CA-125 level of less than the upper limit of normal. Using Next-Generation Sequencing assay, tumor tissue was examined to determine whether DNA contained a deleterious somatic or germline BRCA mutation (tBRCA), in addition to determining the percentage of genomic Loss of Heterozygosity (LOH). Positive Homologous Recombination Deficiency (HRD) status was defined as tBRCA-positive and/or LOH high. The Primary end point was Progression Free Survival in three patient cohorts – all patients, HRD subgroup, and tumor BRCA subgroup.
It was noted that there was a statistically significant improvement in median Progression Free Survival (PFS) for all patients assigned to RUBRACA®, compared with placebo (median PFS 10.8 versus 5.4 months, HR=0.36; P<0.0001). In the HRD subgroup, the median PFS was 13.6 months for those assigned to RUBRACA®, versus 5.4 months for the placebo group (HR=0.32; P<0.0001), and in the tumor BRCA subgroup, the median PFS was 16.6 versus 5.4 months (HR=0.23; P <0.0001), respectively. The most common adverse reactions were fatigue, rash, nausea, vomiting, diarrhea, abdominal discomfort, cytopenias and abnormal liver function studies. Discontinuation due to adverse reactions occurred in 15% of patients receiving RUBRACA®.
It was concluded that RUBRACA® significantly improved Progression Free Survival in patients with platinum-sensitive ovarian cancer who had achieved a response to platinum-based chemotherapy, and could be considered a new standard of care for women with platinum-sensitive ovarian cancer, following a complete or partial response to second-line or later lines of platinum-based chemotherapy. The FDA also concurrently approved the complementary diagnostic test, FoundationFocusTM CDx BRCA LOH for tumor samples, to determine HRD status.
Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Coleman RL, Oza AM, Lorusso D, et al. The Lancet 2017;390:1949-1961
VERZENIO® (Abemaciclib)
The FDA on February 26, 2018 approved VERZENIO® in combination with an Aromatase Inhibitor, as initial endocrine-based therapy for postmenopausal women with Hormone Receptor (HR)-positive, Human Epidermal growth factor Receptor 2 (HER2)-negative advanced or metastatic breast cancer. VERZENIO® is a product of Eli Lilly and Company.
IMFINZI® (Durvalumab)
The FDA on February 16, 2018 approved IMFINZI® for patients with unresectable stage III Non-Small Cell Lung Cancer (NSCLC,) whose disease has not progressed following concurrent platinum-based chemotherapy and radiation therapy. IMFINZI® is a product of AstraZeneca Inc.
FDA Approves 4-Week Dosing Schedule for OPDIVO®
SUMMARY: The FDA on March 6, 2018 approved a supplemental Biologics License Application (sBLA) updating the OPDIVO® (Nivolumab) dosing schedule to include 480 mg infused every four weeks (Q4W) for a majority of approved indications. OPDIVO® is an immune checkpoint PD-1 (Programmed cell Death 1) targeted, fully human, immunoglobulin G4 monoclonal antibody approved by the FDA for multiple tumor types. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Under normal circumstances, Immune checkpoints or gate keepers, inhibit intense immune responses by switching off the T cells of the immune system. They therefore suppress antitumor immunity. OPDIVO® by targeting immune checkpoint PD-1, unleashes the T cells, resulting in T cell proliferation, activation and a therapeutic response.
The clinical pharmacology of OPDIVO® is well established and the clinical data regarding the efficacy and safety of OPDIVO® when administered at 3 mg/kg Q2W (every 2 weeks) across multiple tumor types is well characterized. However, alternative dosing schedules would provide flexibility and other benefits both to patients as well as prescribers.
The authors in this study, using a combination of quantitative clinical pharmacology analyses and safety assessments, evaluated the feasibility of extending the dosing interval of OPDIVO®, and administering it every 4 weeks instead of every 2 weeks. They examined the predicted risk/benefit profile of OPDIVO® 480 mg Q4W compared to 3 mg/kg Q2W by
(1) Comparing OPDIVO® exposures produced by 3 mg/kg Q2W and 480 mg Q4W across tumor types
(2) Evaluating OPDIVO® exposure margins for safety, relative to the well-tolerated dose of 10 mg/kg Q2W
(3) Comparing the predicted risk of experiencing grade 3 adverse events with 480 mg Q4W relative to 3 mg/kg Q2W across the various tumor types for which it is indicated
(4) Comparing the predicted Objective Response Rate (ORR) and Overall Survival with OPDIVO® 480 mg Q4W compared to 3 mg/kg Q2W, in patients with Melanoma, Non Small Cell Lung Cancer (NSCLC), and Renal Cell Carcinoma (RCC).
The researchers noted that among patients with Melanoma, NSCLC, or RCC, there was a less than 1% difference in the predicted probability of achieving a response. The predicted 1 and 2-year survival probabilities were also similar among patients with these tumor types receiving either of the two dose schedules of OPDIVO®, with differences ranging between 0-4.6% at the end of the first year and 1.9-6.9% at the end of second year, across tumor types.
Based on this data, OPDIVO® 480 mg Q4W flat dose option was approved by the FDA for the following indications:
• Metastatic melanoma (monotherapy or monotherapy phase after combination treatment with YERVOY® (Ipilimumab)
• Previously treated metastatic Non Small Cell Lung Cancer
• Advanced Renal Cell Carcinoma following prior Anti-angiogenic therapy
• Previously treated locally advanced or metastatic Urothelial carcinoma following disease progression during or after Platinum-based chemotherapy
• Classical Hodgkin lymphoma following relapse/progression after autologous Hematopoietic Stem Cell Transplantation (HSCT) and Brentuximab vedotin, or three or more lines of systemic therapy that includes autologous HSCT
• Recurrent/metastatic Squamous Cell Carcinoma of the Head and Neck following Platinum-based therapy
• Hepatocellular carcinoma after prior Sorafenib therapy
• Adjuvant therapy for patients with completely resected Melanoma with lymph node involvement or metastatic disease
It was concluded that based on the clinical pharmacology of OPDIVO® and well characterized Exposure-Response relationships for efficacy and safety, the differences in exposures produced by a OPDIVO® schedule of 480 mg Q4W relative to 3 mg/kg Q2W dosing schedule, should not result in clinically meaningful differences in the safety and efficacy of OPDIVO®. This alternate, flexible dosing option may further help tailor patient care. A model-based exposure-response (E-R) assessment of a nivolumab (NIVO) 4-weekly (Q4W) dosing schedule across multiple tumor types [abstract]. Zhao X, Ivaturi V, Gopalakrishnan M, et al. In: Proceedings of the American Association for Cancer Research Annual Meeting 2017; 2017 Apr 1-5; Washington, DC. Philadelphia (PA): AACR; Cancer Res 2017;77(13 Suppl):Abstract CT101. doi:10.1158/1538-7445.AM2017-CT101
CABOMETYX® Improves Overall Survival in Advanced Hepatocelluar Carcinoma
SUMMARY: The American Cancer Society estimates that for 2018, about 42,220 new cases of primary liver cancer will be diagnosed in the US and 30,200 patients will die of their disease. Liver cancer is seen more often in men than in women and the incidence has more than tripled since1980. This increase has been attributed to the higher rate of Hepatitis C virus (HCV) infection among baby boomers (born between 1945 through 1965). Obesity and type II diabetes have also likely contributed to the trend. Other risk factors include alcohol, which increases liver cancer risk by about 10% per drink per day, and tobacco use, which increases liver cancer risk by approximately 50%. HepatoCellular Carcinoma (HCC) is the second most common cause of cancer-related deaths worldwide. NEXAVAR® (Sorafenib) was approved by the FDA in 2007 for the treatment of unresectable HepatoCellular Carcinoma (HCC). Patients with advanced HCC, who progress on NEXAVAR®, have a poor prognosis, with limited treatment options.
CABOMETYX® (Cabozantinib) is an oral, small-molecule Tyrosine Kinase Inhibitor (TKI) which targets the Vascular Endothelial Growth Factor Receptors (VEGFR), and additionally inhibits the action of tyrosine kinases MET and AXL. Increased expression of MET and AXL is associated with tumor progression and development of resistance to VEGFR inhibitors. Previously published studies demonstrated clinical activity of CABOMETYX® in patients with advanced HepatoCellular Carcinoma (HCC).
The CELESTIAL trial is a global, randomized, double-blind, phase III study, which evaluated the benefit of CABOMETYX® in patients with advanced HCC, whose disease progressed on prior treatment with NEXAVAR® or other systemic therapies. NEXAVAR® is considered the standard first line treatment for patients with advanced HCC. In this study, 707 patients were randomized in a 2:1 ratio to receive CABOMETYX® 60 mg daily (N= 470) or placebo (N=237). Eligible patients had an ECOG performance status of 0 or 1, a Child-Pugh score of A, and had progressed on at least one prior systemic therapy for advanced HCC, with 70% having received only prior treatment with NEXAVAR® and 27% having received two prior systemic therapy regimens for advanced HCC.. The median age was 64 years, 38% had Hepatitis B Virus, 24% had Hepatitis C Virus, 78% had ExtraHepatic Spread (EHS), 30% had MacroVascular Invasion (MVI) and 85% had both EHS and MVI. Both treatment groups were well balanced and patients were stratified based on etiology of disease, geographic region, and the presence of EHS and/or MVI. The Primary endpoint was Overall Survival (OS) and Secondary endpoints included Progression Free Survival (PFS) and Objective Response Rate (ORR).
This study met the Primary endpoint at the second planned interim analysis and the median Overall Survival was 10.2 months with CABOMETYX®, compared with 8.0 months with placebo (HR=0.76; P=0.0049) , which meant a 24% reduction in the risk of death. Among patients who received NEXAVAR® alone and received CABOMETYX® as second-line treatment, the median survival was 11.3 months versus 7.2 months with placebo. (HR =0.70). CABOMETYX® also improved PFS compared to placebo and the median PFS was 5.2 months with CABOMETYX® versus 1.9 months with placebo (HR=0.44; P<0.0001). Although the Objective Response Rate was only 4% with CABOMETYX® versus 0.4% with placebo (P=0.0086), stable disease rates however, were doubled (60% vs 33%). The most common grade 3 adverse events in the CABOMETYX® group was hand-foot skin reaction, hypertension, elevated liver enzymes, fatigue and diarrhea.
It was concluded that CABOMETYX® significantly improved Overall Survival and PFS, compared with placebo, in previously treated patients with advanced HCC, and CABOMETYX® represents a new treatment option for this patient group. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: Results from the randomized phase III CELESTIAL trial. Abou-Alfa GK, Meyer T, Cheng A-L, et al. J Clin Oncol 36, 2018 (suppl 4S; abstr 207)
CHEMOTHERAPY INDUCED NAUSEA AND VOMITING (CINV)
Chemotherapy Induced Nausea and Vomiting (CINV) is quite common and occurs in about 80% of patients receiving chemotherapy1,2. It is important to differentiate nausea from vomiting, as more patients experience nausea rather than vomiting3. There are 6 different categories of CINV
1. Acute CINV
2. Delayed CINV
3. Breakthrough CINV
4. Refractory CINV
5. Anticipatory CINV
6. Radiation Induced Nausea and Vomiting
ACUTE CINV: Acute CINV by definition begins within the first 24 hours following chemotherapy administration, with most patients experiencing symptoms within the first four hours of treatment.
DELAYED CINV: This is defined as nausea and vomiting occurring more than 24 hours after chemotherapy administration and can persist for several days4. This is often underestimated, as a third of the patients receiving chemotherapy may experience delayed nausea and vomiting with out prior acute nausea or vomiting.
BREAKTHROUGH CINV: This is defined as nausea and/or vomiting experienced by patients despite prophylactic treatment with antiemetics. These patients require additional intervention with antiemetics to treat their symptoms.
REFRACTORY CINV: Patients on chemotherapy may experience nausea and vomiting following subsequent cycles of chemotherapy administration when prophylaxis/rescue for nausea and vomiting have failed in earlier cycles.
ANTICIPATORY CINV: This is a “learned conditioned response” with psychologic undertones as a result of prior experience of nausea and vomiting with chemotherapy. It usually occurs about 12 hours leading up to chemotherapy administration5,6. The incidence varies from 20%-50%. It is more often seen in younger patients and nausea is more common than vomiting. This entity is difficult to treat and may require psychologic counseling.
RADIATION INDUCED NAUSEA AND VOMITING: This is often experienced by patients undergoing Total Body Irradiation (TBI), a technique that is used prior to bone marrow transplantation7. Patients undergoing radiation to the upper abdomen will also experience nausea and vomiting8.
Pathophysiology of Nausea and Vomiting
The brain controls the multistep pathway that results in vomiting. There are two separate units in the medulla that play a vital role in the causation of nausea and vomiting.
• Chemoreceptor trigger zone (CTZ) also called Area Postrema, present on the floor of the fourth ventricle. This area is very sensitive to chemical stimuli and is easily accessible to emetogenic substances in the general circulation.
• Vomiting Center (Emetic Center) located in the reticular formation of the medulla. This center integrates the emetic response and coordinates the act of vomiting.
The neuroreceptors involved in mediating vomiting include Serotonin (5HT), Dopamine (D2), Neurokinin-1(NK-1), Cannabinoid(CB1), Opiate, Histamine(H1), Corticosteroid and Acetylcholine or Muscarinic(M) receptors. These receptors are located in the vomiting and vestibular centers of the brain9. The most important receptors involved in the emetic response however are Serotonin and Dopamine receptors10. Vomiting is triggered by the Vomiting Center after it receives impulses from CTZ, GI tract, and cerebral cortex and vestibular apparatus in the inner ear. Chemotherapeutic agents and radiation therapy generally induce vomiting by producing free radicals, which in turn act on the enterochromaffin cells, resulting in the release of Serotonin(5-hydroxytryptamine-5HT3). Serotonin activates the serotonin receptors. Activation of the receptors then activates the vagal afferent pathway, which in turn activates the vomiting center and causes an emetic response. Chemotherapeutic agents along with its metabolites also directly stimulate the CTZ by way of general circulation, triggering vomiting through the Vomiting Center.
Classification of Chemotherapeutic agents based on Emetogenicity11
High Risk: More than 90% of the patients experience acute emesis
Moderate Risk: 30-90% of the patients experience acute emesis
Low Risk: 10-30% of the patients experience acute emesis
Minimal Risk: Less than 10% of the patients experience acute emesis
Risk Factors for Nausea and Vomiting
• Emetogenic potential of the chemotherapy agent used
• Younger age
• Female gender
• History of motion sickness
• Alcohol consumption
Classification of Antiemetics
Serotonin (5-HT3) Receptor Antagonists
ZOFRAN ® (Ondansetron): This agent is the first in its class approved by the FDA in 1991 for the treatment of CINV. This first generation 5-HT3 receptor antagonist is available as an IV preparation, sublingual/chewable tablet, oral tablet and oral solution. This agent has a shorter half life than KYTRIL® and ALOXI®.
KYTRIL® (Granisetron): This first generation 5-HT3 receptor antagonist provides 24-hour protection against chemotherapy induced nausea and vomiting. It is available as a single dose Injection as well as tablets or oral solution
ALOXI ® (Palonosetron): This second generation 5-HT3 antagonist has a 100 fold higher binding affinity to 5-HT3 receptor than other 5-HT3 receptor antagonists12. This agent is effective in preventing both acute and delayed onset nausea and vomiting. This agent has to be given IV and oral administration is not feasible due to poor bioavailability, unlike the first generation 5-HT3 antagonists.
Neurokinin-1 (NK-1) Receptor Antagonist
EMEND® (Aprepitant): This agent selectively blocks the binding of substance P to the NK-1 receptor in the central nervous system and thereby complements the antiemetic activity of 5-HT3 receptor antagonists by virtue of its different mechanism of action. It is available as tablets as well as parenteral preparation. This agent can interact with several other drugs more, so when given orally because of first-pass metabolism13. It is therefore important to check the package insert for drug interactions.
Dopamine Receptor Antagonists
These agents antagonize dopamine (D2)-receptors which are involved in the emetic signaling through the chemoreceptor trigger zone. Dopamine receptor antagonists also counteract dopamine receptors in the stomach, implicated in decreasing stomach motility during nausea and vomiting. The three main classes of dopamine receptor antagonists are phenothiazines, butyrophenones, and benzamides.
Phenothiazines – PHENERGAN® (promethazine) belongs to this class. Low doses of phenothiazines antagonize interaction of dopamine with D2-receptors and thus exert an antiemetic effect.
Benzamides – Are strong central and peripheral D2-antagonists. They exert antiemetic effects by increasing lower esophageal sphincter tone and decreasing transit time through the upper gastrointestinal tract. REGLAN® (metoclopramide) belongs to this class.
Butyrophenones
HALDOL® (Haloperidol) belongs to this group of agents.
H1 Receptor Antagonists
Antihistamines antagonize the H1 receptors and inhibit the action of histamine. They also affect the vestibular system, decreasing stimulation of the vomiting center. Further, they also exhibit activity by inhibiting the muscarinic receptor. However, second generation antihistamines such as CLARITIN® (Loratidine), ALLEGRA® (Fexofenadine) and ZYRTEC® (Cetirizine) do not cross the blood brain barrier, and as such do not cause drowsiness and cannot be used as antiemetics. Some examples of H1 receptor antagonists include
• DRAMAMINE® (Dimenhydrinate)
• ANTIVERT® (Meclizine)
• BENADRYL® (Diphenhydramine)
Muscarinic Receptor Antagonists
These agents are good for motion sickness. They antagonize the acetylcholine receptors in the brain. Scopolamine transdermal belongs to this class
Cannabinoids Receptor Antagonists
These agents antagonize the CB1 receptors in the brain. The two drugs in this class
• MARINOL® (Dronabinol)
• CESAMET® (Nabilone)
Corticosteroids
The mechanism of action not clear. It may be related to the inhibition of arachidonic acid release. The agents in this class include
• DECADRON® (Dexamethasone)
• SOLUMEDROL® (Methylprednisolone)
Benzodiazepines
These agents are sometimes effective for anticipatory nausea and vomiting associated with cancer therapy. It may also be useful for vestibular disorders.
• VALIUM® (Diazepam)
• ATIVAN® (Lorazepam)
• XANAX®: (Alprazolam) Miscellaneous
• TIGAN® (Trimethobenzamide)
General Principles of treating Nausea and Vomiting
1) Prevention is better than cure. Aggressively preventing nausea and vomiting will not only improve patients quality of life but will also decrease the incidence of anticipatory nausea and vomiting.
2) The addition of DECADRON® to 5-HT3 and NK-1 receptor antagonists improves the efficacy of the antiemetic regimen.
3) The choice of an antiemetic regimen should be based on patient risk factors as well as emetogenic potential of a given chemotherapy regimen.
4) The toxicity of a given antiemetic agent and potential drug interactions should be taken into consideration before prescribing.
5) Breakthrough emesis should be aggressively managed with round the clock dosing rather than PRN dosing and a drug from a different class should be considered for treatment of this entity. Also consider rectal or IV route of administration rather than PO route.
6) Ensure patients are well hydrated and electrolyte imbalances are promptly addressed.
7) Think “outside the box” when addressing nausea and vomiting in cancer patients. Do not overlook bowel obstruction, brain metastases, uremia, nausea from pain meds such as opiates, chemo or tumor related gastroparesis and anticipatory nausea and vomiting.
8) Anticipatory nausea and vomiting can be difficult to treat and benzodiazepines (ATIVAN®, XANAX®) along with behavioral therapy may be beneficial.
Reference List
1. Morran C, Smith DC, Anderson DA, McArdle CS. Incidence of nausea and vomiting with cytotoxic chemotherapy: a prospective randomised trial of antiemetics. Br Med J 1979; 1(6174):1323-1324.
2. Jenns K. Importance of nausea. Cancer Nurs 1994; 17(6):488-493.
3. Hickok JT, Roscoe JA, Morrow GR et al. 5-Hydroxytryptamine-receptor antagonists versus prochlorperazine for control of delayed nausea caused by doxorubicin: a URCC CCOP randomised controlled trial. Lancet Oncol 2005; 6(10):765-772.
4. Kris MG, Gralla RJ, Clark RA et al. Incidence, course, and severity of delayed nausea and vomiting following the administration of high-dose cisplatin. J Clin Oncol 1985; 3(10):1379-1384.
5. Moher D, Arthur AZ, Pater JL. Anticipatory nausea and/or vomiting. Cancer Treat Rev 1984; 11(3):257-264.
6. Jacobsen PB, Redd WH. The development and management of chemotherapy-related anticipatory nausea and vomiting. Cancer Invest 1988; 6(3):329-336.
7. Kris MG, Hesketh PJ, Somerfield MR et al. American Society of Clinical Oncology guideline for antiemetics in oncology: update 2006. J Clin Oncol 2006; 24(18):2932-2947.
8. Harding RK. Prodromal effects of radiation: pathways, models, and protection by antiemetics. Pharmacol Ther 1988; 39(1-3):335-345.
9. Dodds LJ. The control of cancer chemotherapy-induced nausea and vomiting. J Clin Hosp Pharm 1985; 10(2):143-166.
10. BORISON HL, WANG SC. Physiology and pharmacology of vomiting. Pharmacol Rev 1953; 5(2):193-230.
11. Grunberg SM, Osoba D, Hesketh PJ et al. Evaluation of new antiemetic agents and definition of antineoplastic agent emetogenicity–an update. Support Care Cancer 2005; 13(2):80-84.
12. Grunberg SM, Koeller JM. Palonosetron: a unique 5-HT3-receptor antagonist for the prevention of chemotherapy-induced emesis. Expert Opin Pharmacother 2003; 4(12):2297-2303.
13. Shadle CR, Lee Y, Majumdar AK et al. Evaluation of potential inductive effects of aprepitant on cytochrome P450 3A4 and 2C9 activity. J Clin Pharmacol 2004; 44(3):215-223.